Targeted protein degradation (TPD) using small molecules termed as molecular glues (MGs) is potential to explore drug discovery and development for treating diseases such as cancer, inflammatory and neurodegenerative diseases. Targeting protein-protein interactions (PPIs), important for regulation of biological systems and development of disease states, via small molecules is a classical approach for drug discovery. The majority of this trend is focussed on inhibition of the activity of proteins/enzymes. Recently, instead of PPI inhibition, selective degradation followed by the elimination of disease causing proteins has brought great attention to generate innovative drug entity. Protein degradation includes two significant advantages over protein inhibition in drug discovery. First, the targeted degradation is a catalytic process associated with transient binding and dissociating after promoting polyubiquitination of the disease-causing protein, and a single degrader can destroy many copies of a pathogenic protein. Second, degraders reduce all functions of protein whilst protein inhibitors block the active site of a pathogenic protein providing high sensitivity to drug-resistant targets.
Ubiquitin, found in almost all eukaryotic organisms, is a firmly conserved protein, whereas the ubiquitin-proteasome system (UPS) is a tightly regulated mechanism for intracellular protein degradation and maintaining protein homeostasis carried out by a complex cascade of enzymes that result in ubiquitination of the protein of interest (POI). The E3 ligases are critical components of the ubiquitination cascade. So far, only a few of E3 ligases out of the >600 E3 ubiquitin ligases encoded by the human genome have been exploited for TPD application, for example, cereblon (CRBN), VHL, MDM2, DDB1, DCAF15, and SCF βTRCP.
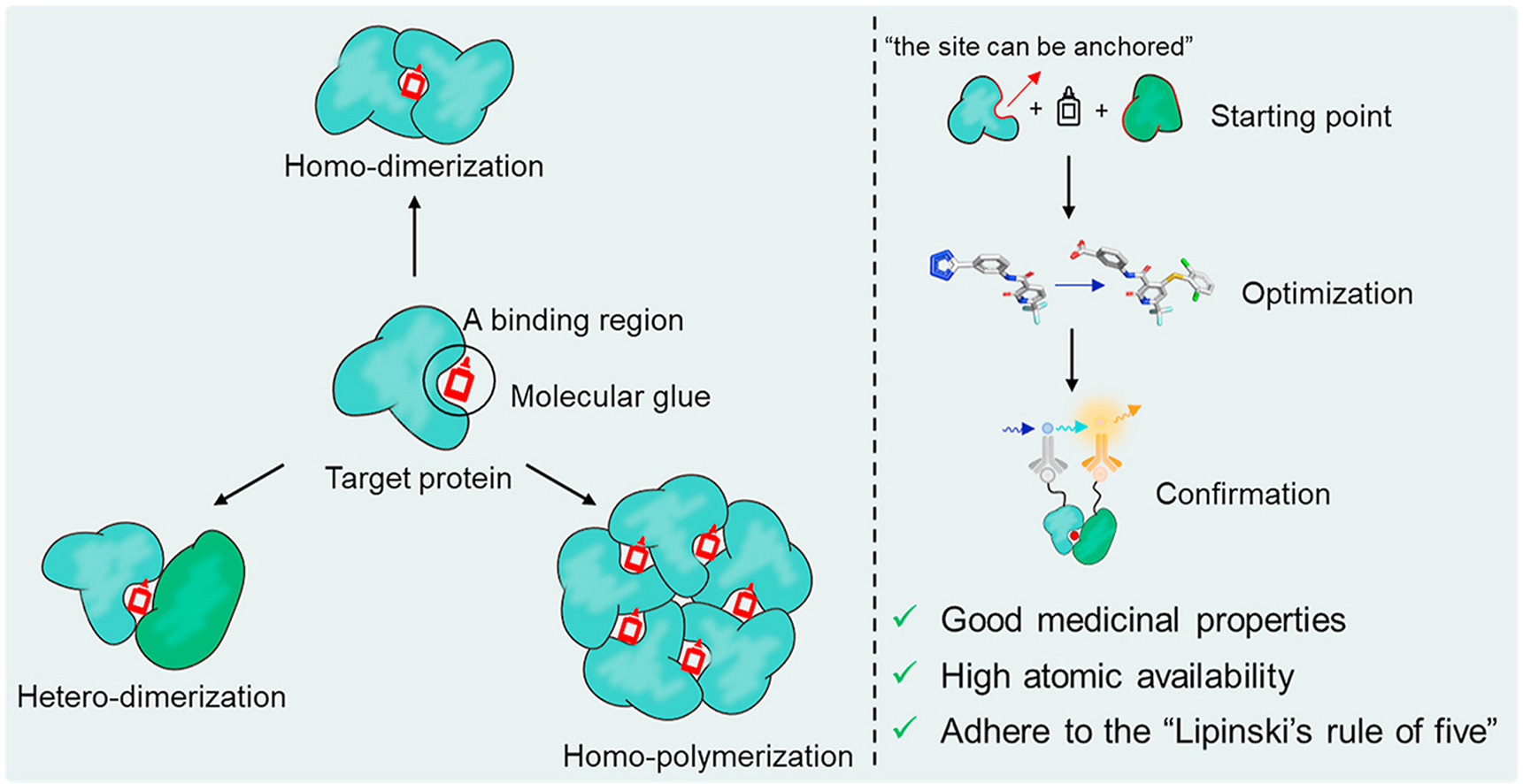
The two leading protein-degrading approaches by small molecules are heterobifunctional proteolysis-targeting chimeras (PROTACs) and MGs. They have different modes of action and structural features (Figure-1) [1]. MGs, natural or synthetically prepared, stabilize interactions between E3 ubiquitin ligases and POI by biological functions, such as signal transduction, transcription, chromatin regulation, and protein folding and localization to convert the target protein into a “neo-substrate” for an E3 ligase leading to its degradation. In contrast, PROTACs create bond between POI and E3 ubiquitin ligase connecting via a linker to form a ternary complex leading to polyubiquitination and degradation. MGs have advantages over PROTACs such as MGs are smaller molecules, follow Lipinski’s rule of 5, have higher cell permeability, favourable PK profile, lower affinity for ligand or protein etc. compared to PROTACs. Molecular mechanisms of PROTACs are predictable and can reasonably be designed according to the binding mode of ligands to target proteins. However, MGs have lack of systematic discovery methods and rational design strategies.
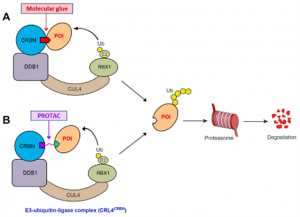
Figure-1: Graphical presentation of the degradation of a POI by the UPS using a MG (A) or PROTAC (B)
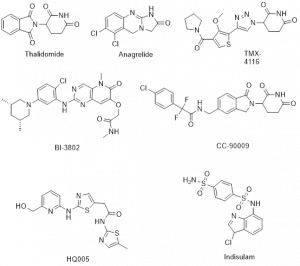
Figure-2: Example of several molecular glues that demonstrate potential drug candidates via targeted protein degradation
In drug discovery, the structure based drug design (SBDD) is the effective, rapid, specific and economical process for hit to lead generation and lead optimization. The protein crystal structure, in silico modelling and computational docking analysis provide significant support for SBDD. In recent years, TPD is the most powerful SBDD strategy. In the last three decades, there have been rapid advances in the identification of targeted protein degraders for therapeutic purposes. In 2001, there was a successful report of the degradation of a cancer-associated protein along with the in vitro proof-of-concept study. The first small-molecule PROTAC was reported in 2008, which included an androgen receptor (AR) degrader, a ligand nutlin-3 for recruiting of MDM2 and a PEG-based linker. Particularly over the last two decades, many exemplary proteins were successfully targeted for E3 ligase degradation [1,2]. According to the CAS content collection, there are >1000 publications including articles and patents connected to the TPD. Specifically in the last few years, significant efforts have given by academia and drug discovery institutions in many countries mainly in USA, China, UK, Japan, Germany, France and India. The TPD-related research was highly dominated by the drug discovery companies. Among them, Ambagon Therapeutics (USA), Bristol Myers Squibb (USA), Ranok Therapeutics (China), Novartis (Switzerland), Monte Rosa Therapeutics (USA) and C4 Therapeutics (USA) are notable. They are trying to make the molecular glue degrader drugs, which are in progression to the clinic.
Progression of drug discovery via targeted protein degradation with molecular glues
The majority of the classical molecular glues have been discovered serendipitously, but exploration of their systematic discovery and rational design are progressing rapidly. Thalidomide and its analogues lenalidomide and pomalidomide are FDA approved immunomodulatory imide drugs (IMiDs) [1] since late 1950s, before their functional mechanisms were elucidated. Interestingly, they were discovered serendipitously as molecular glue to facilitate the interactions between E3 ligase cereblon (CRBN) and POI resulting in subsequent ubiquitination and protein degradation. According to an initial report, transcription factors protein IKZF1 and IKZF3 were ubiquitinated by IMiDs-induced CRL4CRBN and degraded by proteasome leading to the antitumor and immunomodulatory properties of IMiDs. The conserved glutarimide ring of IMiDs is liable to bind to CRBN via hydrogen bonds and van der Waals contact. Recently, many thalidomide analogues and compounds having the core CRBN binding pharmacophore glutarimide ring have been identified as MGs reported in journals and patents to enhance potency, selectivity and drug-like properties. Preclinical and phase I/II clinical trials for the treatment of different type of cancers such as myeloma, lymphoma and melanoma with several drug candidates via degrading transcription factors IKZF1 and IKZF3 are in evaluation [1]. Few natural compounds have also been found to function as molecular glues [1].
Recently, different approaches have discovered molecular glue degraders of structurally distinct cyclin K and CDK12, which are promising drug targets to treat human cancers. The optimization of several MG drug candidates, which degrades cyclin K and CDK12 via distinct mechanism, is advancing. For example, HQ005 [1] is a leading drug candidate gluing DDB1 to CDK12 for cyclin K degradation. Another multifunctional protein of the CK1 protein family is casein kinase 1α (CK1α), which regulates signalling pathways involving autoimmune diseases, neurodegenerative diseases, and cancer. Few triazole derivatives (eg, TMX-4116) [1] act as molecular glue to degrade CK1α and are under therapeutic application for treatment of multiple myeloma.
Some other potential targeted proteins are G1 to S phase transition protein 1 (GSPT1), Sal-like protein 4 (SALL4), RNA-binding motif protein 39 (RBM39), β-catenin and BCL6 protein, whose degradations by MGs have been reported. The CC-90009 [3] is a highly selective and potent CRBN-mediated protein degrader, which is currently under phase 1 clinical trial for treating acute myeloid leukemia. SALL4 is responsible for a variety of cancers such as hepatocellular carcinoma, breast, lung and colorectal cancer. No drug is available for targeting SALL4. Thalidomide and its derivatives have been reported to induce SALL4 degradation, however it is still a challenging area for the medicinal chemists to develop an anticancer drug candidate through MG mediated SALL4 degradation. RBM39 is responsible for the antitumor activity that involves transcriptional regulation, alternative splicing and protein translation. Few aryl sulphonamides, for instance such as Indisulam [1], act to adhesive RBM39 with DCAF15 followed by sulfonamide-dependent degradation, whose clinical trials have been evaluated as antitumor drug candidates. The MG induced interaction of the oncogenic transcription factor, β-catenin with its cognate E3 ligase and SKP1β-TrCP has been reported to associate with the scope of anticancer drug. BI-3802 [1] and similar compounds bind bric-a-brac (BTB) domain of BCL6 with SIAH1 E3 ligase resulting in proteasomal degradation with high potency for diffusing large B-cell lymphoma (DLBCL). Anagrelide is a drug to reduce blood platelet count in humans, which had unclear mode of action. Very recently, anagrelide [4] has been identified as a molecular glue to stabilize a complex between cAMP PDE3A and SLFN12.
In conclusion, the area of drug discovery through molecular glues mediated targeted protein degradation is advancing rapidly to deliver specific medicines, especially anticancer drugs. In recent years, a lot of work has been undertaken to design, synthesize, study the biological mechanism and evaluate the activities of molecular glues. Despite difficulties to the identifying and rationally designing, molecular glues have been discovered and optimized. The clinical studies of several drug candidates, originated from molecular glues mediated proximity-induced targeted protein degradation, are in progression.
References
[1] Janet M. Sasso, Rumiana Tenchov, DaSheng Wang, et al; Biochem. 2023, 62, 601-623.
[2] Iacovos N. Michaelides, Gavin W. Collie; J. Med. Chem. 2023, 66, 3173–3194.
[3] Joshua D. Hansen, Matthew Correa, Matt Alexander, et al; J. Med. Chem. 2021, 64, 1835- 1843.
[4] Nicholas A. Meanwell; ACS Med. Chem. Lett. 2023 14, 350-361.
