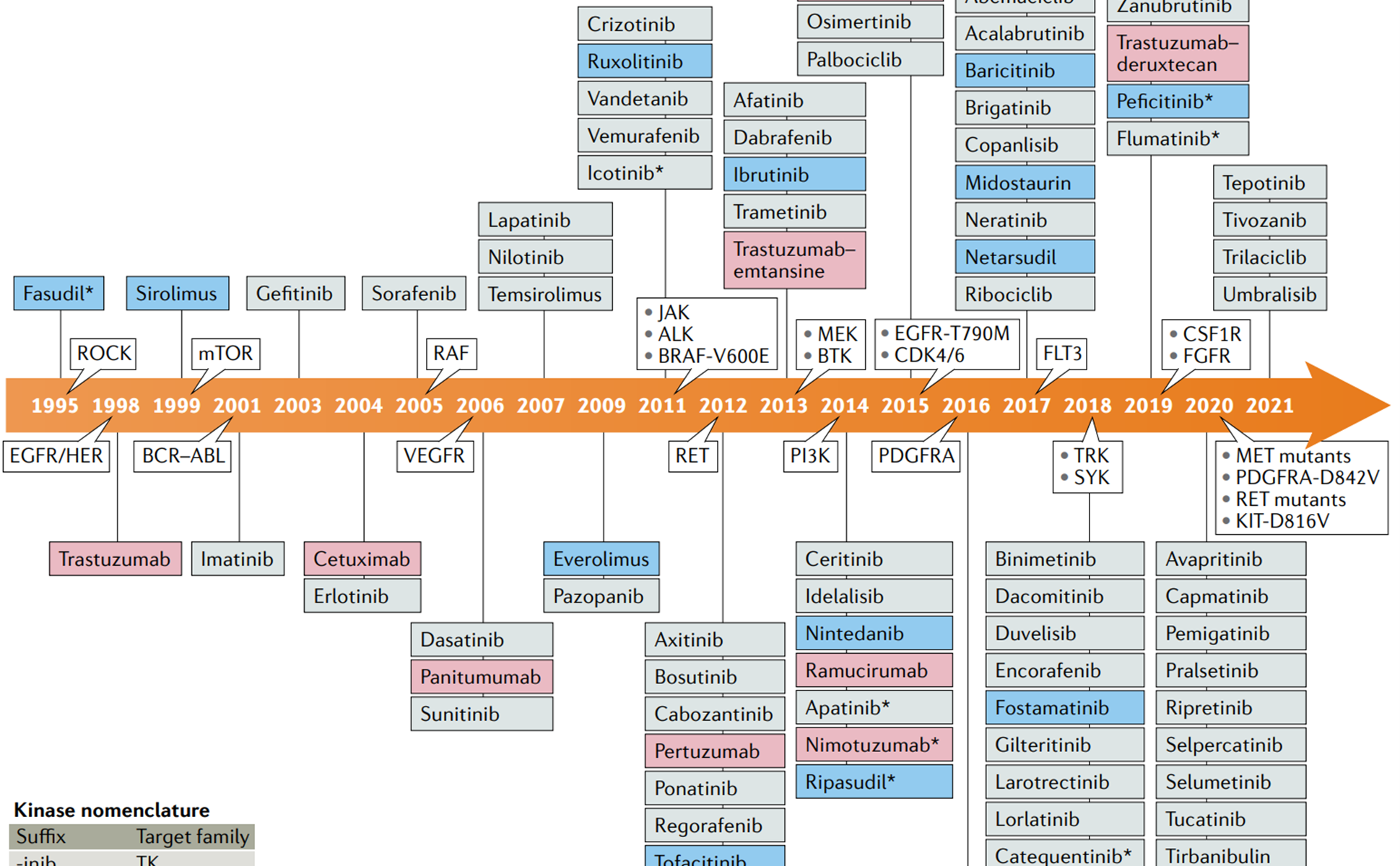
Messenger RNA (mRNA) possesses extensive clinical potential for a range of therapeutic applications including vaccination, protein replacement, gene editing, immunotherapies, and tissue regeneration. The remarkable clinical utility of mRNA, especially in the context of vaccines, is exemplified by its ability to mitigate the ongoing SARS-CoV-2 pandemic. It is important to note that mRNA is a single-stranded RNA variant involved in protein synthesis and synthesized from a DNA template during transcription (Figure 1) [1]. Its crucial role lies in transporting protein information from the DNA in a cell’s nucleus to the cytoplasm, where the protein-making machinery interprets the mRNA sequence and translates each three-base codon into its corresponding amino acid, forming a growing protein chain. However, mRNA faces challenges such as susceptibility to degradation by serum endonucleases and limited cellular entry due to its negative charge. To maximize therapeutic benefits, it is imperative to protect mRNA molecules from degradation and precisely deliver them to the target cells for the desired protein production. Consequently, diverse strategies have arisen to engineer innovative mRNA delivery vehicles that promote effective transfection while minimizing toxicity.
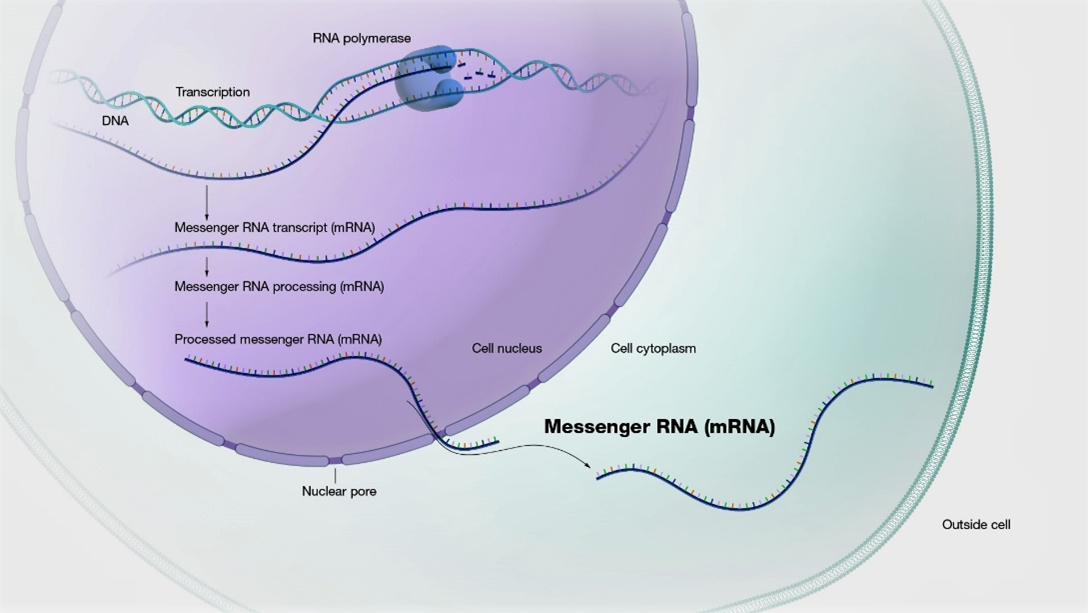
Figure 1: mRNA is made from a DNA template during the process of transcription.
The transport of naked mRNA across the cell membrane is challenging, leading to the development of various delivery carriers such as lipid-based nanoparticles, polymer-based nanoparticles, peptide-based vehicles, and protein-mRNA complexes. Among them, lipid nanoparticles (LNPs) have emerged as the most advanced vectors. LNPs possess numerous advantages over viral vectors for gene therapy applications. These include their moderate or lower immunogenicity, ability to accommodate a large payload, ease of production, and excellent scalability. Additionally, LNPs can be tailored to specific cell targets and disease-related applications by adjusting the lipid-to-mRNA ratio. Upon administration, LNPs are internalized by host cells. Consequently, the encapsulated mRNA cargo is delivered intracellularly to the cytosol, where the mRNA sequences are subsequently translated into targeted proteins by the host cell’s ribosome. The utilization of LNPs in COVID-19 mRNA vaccines is gaining significant attention due to their vital role in preserving and effectively delivering the mRNA payload to the targeted cells. Particularly, mRNA-LNP vaccines against COVID-19 are currently in clinical use, representing a groundbreaking approach to mRNA-based treatment.
LNPs are typically spherical structures composed of a lipid bilayer and an enclosed aqueous compartment. These LNPs possess a sophisticated internal arrangement that imparts them with superior physical stability, owing to their distinctive structural characteristics. Traditional LNPs have generally comprised four key elements: an ionizable or cationic lipid for nucleic acid binding and facilitating escape from endosomes, a polyethylene glycol (PEG) lipid to enhance colloidal stability and reduce clearance by the reticuloendothelial system, a cholesterol component to support LNP stability, and an amphipathic phospholipid to encourage fusion with cell and endosomal membranes. While these traditional LNPs have demonstrated safety and efficacy, their administration has been limited to intramuscular use. This limitation primarily arises from their physiochemical resemblance to very-low-density lipoprotein and their tendency to adsorb apolipoprotein E in blood plasma, leading to accumulation in the liver and uptake into hepatocytes via the low-density lipoprotein receptor.
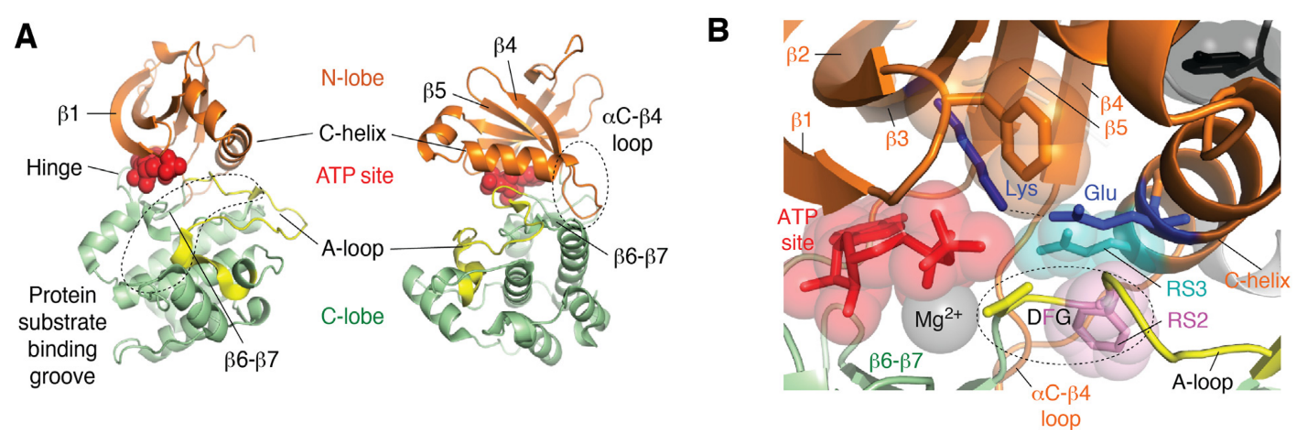
In response to the aforementioned challenge, a methodology known as selective organ targeting (SORT) has been developed. This methodology involves the systematic engineering of nanoparticles to accurately deliver therapeutic molecules to both hepatic and extrahepatic tissues. The mechanistic study discovered that the biophysical class of SORT molecule generates a distinct protein corona when incorporated into the LNP. This corona plays a crucial role in determining the site of mRNA delivery within the body. The objective of organ targeting is not to deliver all doses to the target organs, but rather to deliver a sufficient dose to achieve the desired biological effects while limiting off-target cumulative toxicity. Upon entering the systemic circulation, a majority of the nanoparticles accumulate in the liver. However, targeting organs other than the liver has remained an unresolved issue for a significant period. By incorporating an auxiliary component called selective organ targeting molecules into lipid nanoparticles, it is possible to target selectively the liver, spleen, lungs, and other organs (Figure 2) [2].
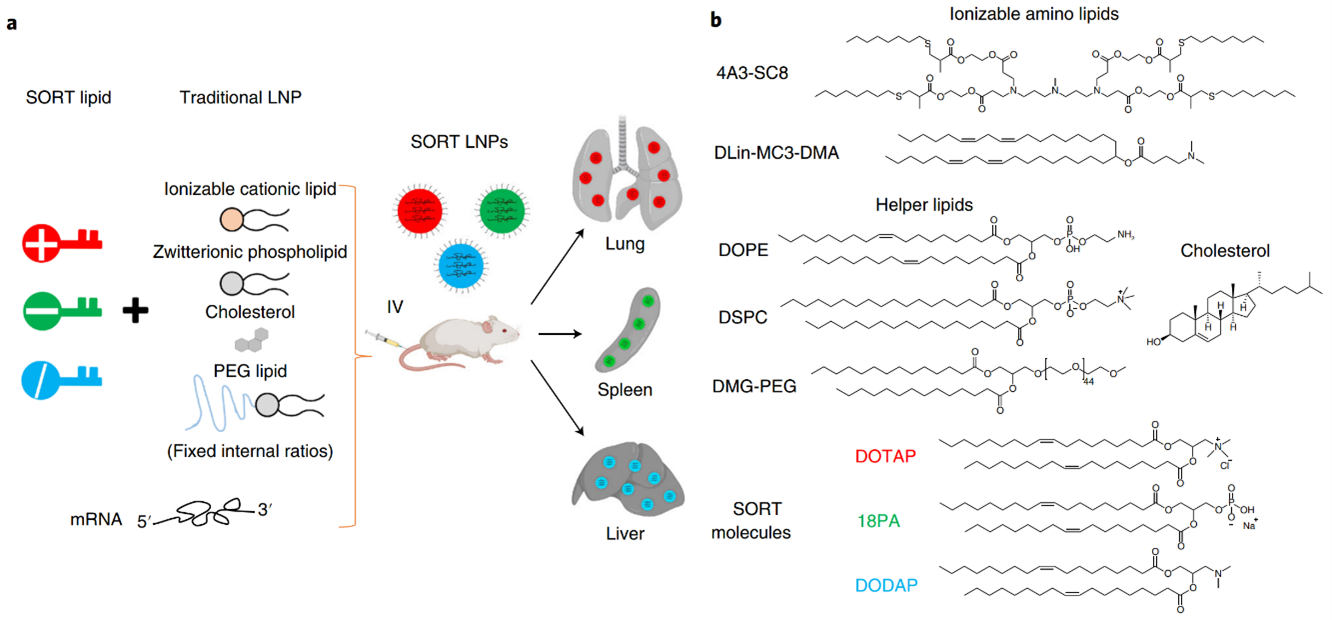
Figure 2: a) The addition of a SORT lipid molecule to typical four-component LNPs alters the in vivo delivery profile of the resultant five-component SORT LNPs, allowing for tissue-specific delivery of mRNA to the liver, lungs, and spleen of mice after IV injections. b) Chemical structures of the lipids used in the protocol, including the ionizable cationic lipids 4A3-SC8 and DLin-MC3-DMA (MC3), the phospholipids DOPE and DSPC, cholesterol, DMG-PEG and the SORT molecules DOTAP, 18PA and DODAP.
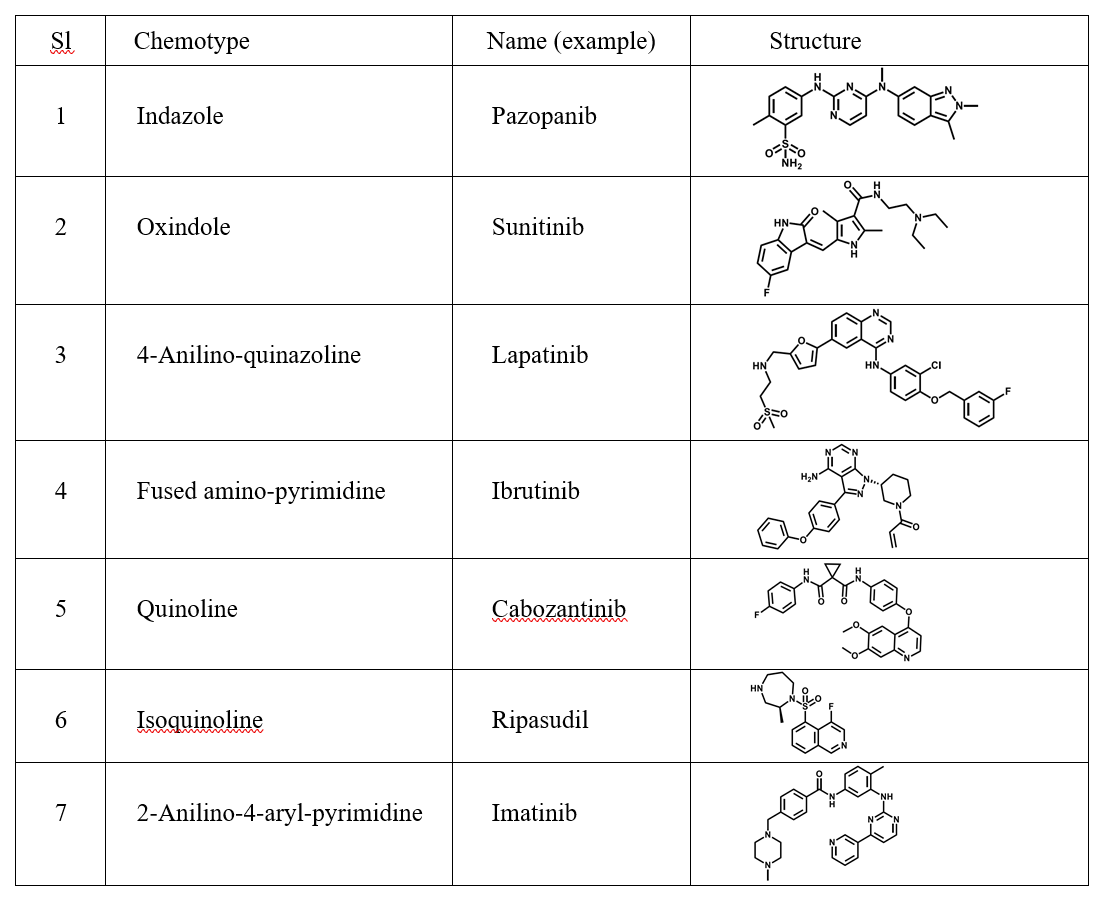
Designing nanoparticles that target specific organs is crucial for effective drug delivery. Each organ, including the brain, heart, liver, spleen, lung, kidney, eye, bone, and bowel, possesses unique characteristics. Therefore, tailoring nanoparticles to match these organ-specific characteristics plays a pivotal role in developing organ-targeting nanoparticles. It is generally necessary to employ a comprehensive array of targeting strategies to achieve the most effective organ-targeting outcomes. These strategies include passive targeting through the utilization of unmodified nanoparticles’ surface properties; active targeting achieved by modifying nanoparticles’ surface to facilitate the transportation of therapeutic molecules to the intended location for therapeutic purposes; endogenous targeting by designing nanoparticles that bind to specific plasma proteins in the blood; and stimuli-responsive targeting that leverages the distinct properties of nanomaterials to switch under specific conditions (Figure 3) [3]. There have been significant achievements of delivering mRNA to various organs with the use of LNPs [4].
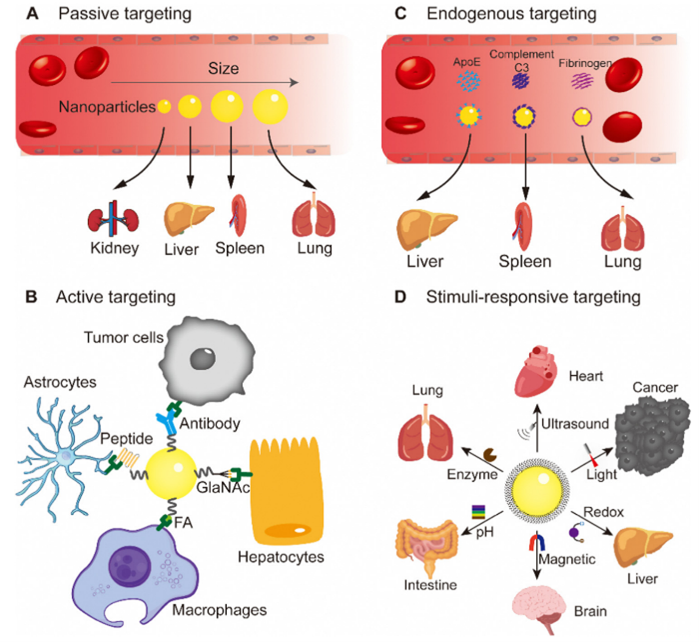
Figure 3: Targeting strategies using nanoparticles: (A) passive targeting, (B) active targeting, (C) endogenous targeting, and (D) stimuli-responsive targeting.
Engineering the formulation of SORT-LNPs allows them to enhance their selectivity and potency for mRNA delivery towards various organs. For example, the utilization of a permanently cationic quaternary ammonium lipid as a SORT molecule in LNPs enables the lung-specific mRNA delivery and genome editing in both pulmonary endothelial and epithelial cells. The potential for clinical translation of selective organ-targeting nanoparticles is enormous. At present, there is a significant amount of ongoing research focused on the utilization of SORT LNPs for the efficient delivery of mRNA, with the ultimate goal of delivering targeted therapeutics. Significant progress has already been made in ongoing research on SORT LNPs-mediated tissue-specific mRNA delivery. Notable achievements include mRNA delivery to lung for lung cancer and pulmonary lymphangioleiomyomatosis, to spleen for cancer, to liver for polyneuropathy, to eye for inherited retinal degenerations, and to bowel for inflammatory bowel diseases [3,4].
References
[1] https://www.genome.gov/genetics-glossary/messenger-rna, NIH.
[2] Xu Wang, Shuai Liu, Yehui Sun, et al; Nat. protoc. 2023, 18, 265.
[3] Jian Li and Hai Wang; Nanoscale Horiz. 2023, 8, 1155.
[4] Kelly Godbout and Jacques P. Tremblay; Pharmaceutics 2022, 14, 2129.