Precision medicine vs traditional medicine
Precision medicine, also known as personalized medicine (PM), is a specialized form of medicine that utilizes information about an individual’s genes, proteins, environment, and lifestyle to prevent, diagnose, and treat diseases. This approach allows doctors and researchers to more accurately predict which treatments will be most effective for a specific patient. In contrast, traditional medicine focuses on developing treatments for large groups of people with the same illness, without considering individual variations.
Cancer
Cancer is a serious health concern that presents a significant risk to human health. Research in the field of oncology has always prioritized understanding the origins, diagnosis, and treatment of cancer. Cancer develops when abnormal cells proliferate uncontrollably, disrupting the normal function of the body. Unlike healthy cells, cancer cells do not follow the normal process of growth, division, and cell death. There are various types of cancer, including lung, colorectal, stomach, liver, prostate, breast, cervical, and thyroid carcinoma, each with unique characteristics related to the affected organ or tissue, behavior of cancer cells, and potential for metastasis. Despite advancements in cancer treatment, it remains the second leading cause of death globally, resulting in approximately 10 million deaths annually, as reported by the International Agency for Research on Cancer (IARC) [1].
Gene and cancer
Cancer is a genetic disease. Genes are comprised of sequences of DNA and are organized, sequentially, at specific locations on chromosomes within the nucleus of cells. Genes provide instructions for protein synthesis, which is essential for cell function. Every gene contains the code to produce a particular protein, each of which has a specific role within the cell. Cancer arises from genetic alterations of some kind. Cancer cells are abnormal variants of normal cells, indicating that a genetic change occurred in a normal cell to transform it into a cancerous cell. For instance, genes that typically regulate cell growth could be deactivated, or genes that control cell division might be constantly activated. Multiple gene mutations are typically required for cancer to develop. The majority of gene alterations are due to mutations, which can impair gene function. Genes with cancer-associated mutations are often referred to as cancer genes. While most mutations occur during an individual’s lifetime, some can be inherited.
Precision medicine in cancer
Precision medicine in cancer treatment involves customizing therapies based on an individual patient’s genetic makeup, lifestyle, and the specific features of their tumor and its surrounding environment. This personalized approach offers an alternative to standard treatments like chemotherapy and radiation, which may not be effective for all patients and can cause harm to healthy tissues. Due to the diverse nature of cancer, with numerous subtypes based on molecular characteristics, clinicians rely on precision medicine to identify the most suitable therapies by analyzing genetic mutations and other molecular features of the tumor, often through techniques like next-generation sequencing. Precision medicine is utilized in certain cancers to determine the most effective tests and treatments. Doctors may use precision medicine to assess cancer risk, detect cancers early, accurately diagnose specific types of cancer, select the best treatment options, and evaluate treatment effectiveness.
How does precision medicine work in cancer?
Precision drugs target specific genetic alterations that drive normal cells to become cancerous. These drugs also address abnormalities in proteins that have changed due to cancer. The goal of treatment is to target the unique characteristics that make cancer cells malignant. Utilizing molecular profiling to analyze a patient’s genetic and immune profile helps match the characteristics of the tumor to the most effective drugs. Molecular profiling involves studying tissue, blood, or fluid samples in a laboratory to identify abnormalities in genes, cell proteins, and specific tumor markers. This process incorporates advanced bioinformatics and analysis of DNA, RNA, proteins, and immune factors. Additionally, environmental and lifestyle factors play a significant role in how an individual’s disease responds to specific treatments.
Types of cancer where precision medicine is utilized
Some of the more prevalent cancers where precision medicine is currently being utilized include: Colorectal cancer, Breast cancer, Lung cancer, Certain types of leukemia, Certain types of lymphoma, Melanoma, Esophageal cancer, Stomach cancer, Ovarian cancer, Thyroid cancer.
Landmark Discoveries in Precision Oncology
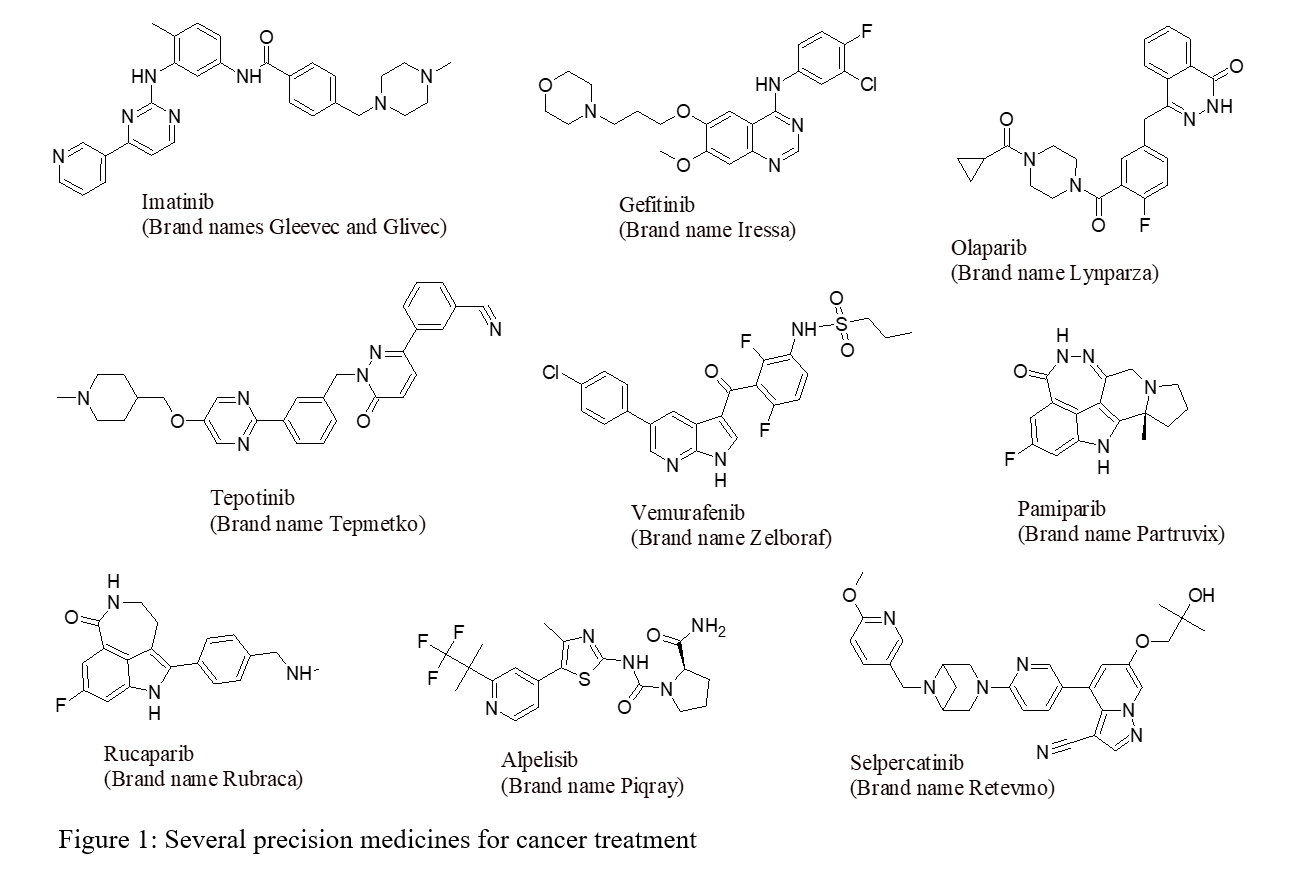
The field of precision oncology has made significant advancements since the discovery of the coupling of estrogen-receptor expression to certain types of breast cancer in the 1970s [2]. Understanding of the biological mechanisms underlying tumor development and patient responses to treatment has grown exponentially, leading to the development of a wide range of targeted therapies. Various strategies have been explored to discover precision medicine for cancer, including the inhibition/blockade of receptor tyrosine kinases, specific targeting of KRAS mutations [3], synthetic lethal approaches targeting homologous recombination deficient cancers with PARP inhibitors [4], biomarker-driven enhancement of response rates to immune checkpoint blockade [3], iron-dependent programmed cell death therapies (Ferroptosis) [5], proteolysis targeting chimeras (PROTACs) [6], and CAR-T cell therapies [3]. Several precision anticancer drugs with structure and brand name are presented in Figure 1.
This new era of precision medicine has resulted in the approval of several new cancer therapeutics every year for biomarker-defined subsets of patients. Recent approvals for biomarker-specific solid tumor treatments have demonstrated the rapid progress in biomarker-driven treatment indications between April 2019 and April 2021 in the United States (FDA) and EU (EMA) ([Figure 2) [7]. These approvals highlight the increasing availability of biomarker-driven treatment options for cancer patients. Approvals related to other biomarker testing methods, such as immunohistochemistry assays, are not included in this overview.
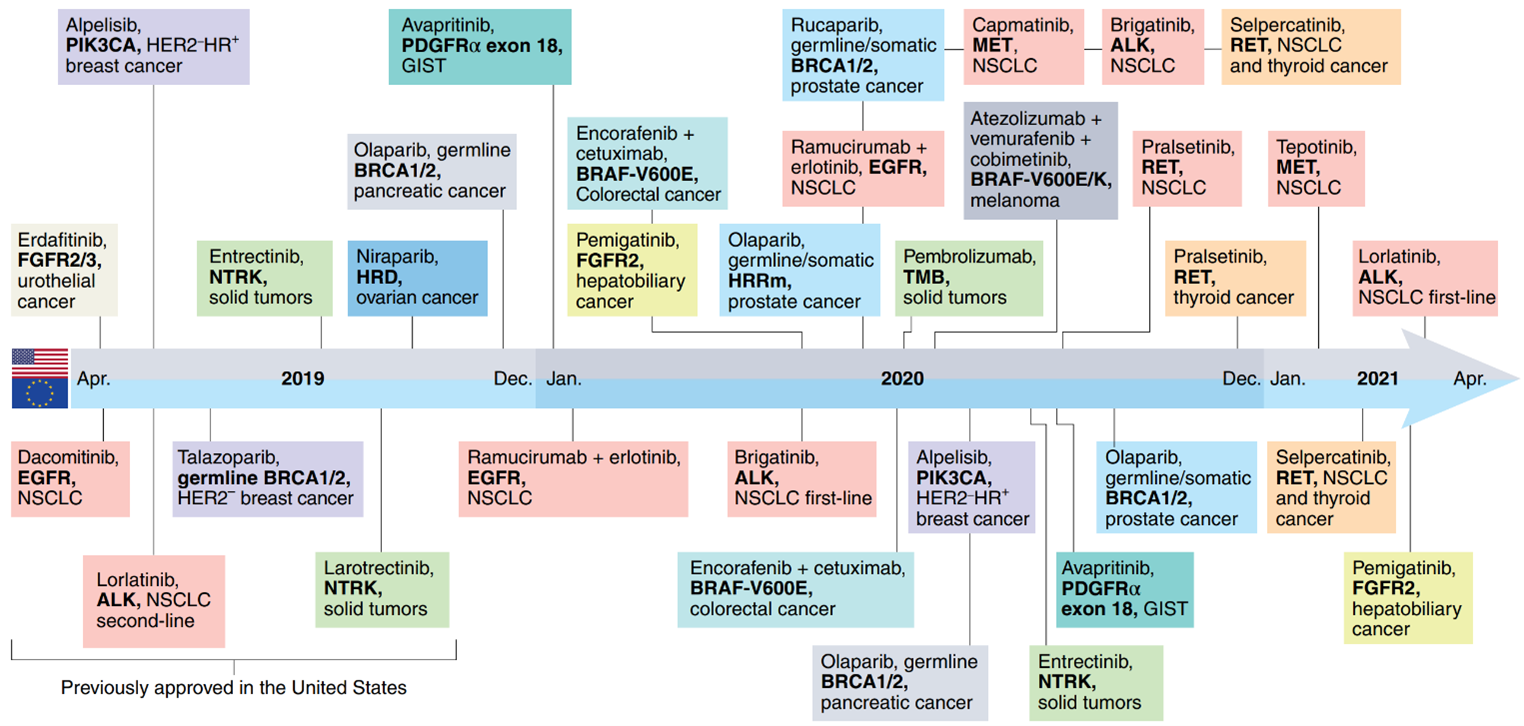
Figure 2: Genomic biomarker-driven drug approvals by FDA (top half) and EMA (EU) (bottom half) in 2019-2021
Challenges in the implementation of precision oncology
Precision oncology, a rapidly growing field in cancer treatment, is attracting significant interest and investment. However, there is a pressing need for empirical evidence to demonstrate its effectiveness compared to traditional therapies. Several challenges hinder the widespread adoption of precision oncology, including the limited availability of molecular-targeted therapies, the high cost of medications, and the potential risks associated with certain treatments. Tumor heterogeneity and ongoing tumor evolution further complicate treatment decisions, as genetic characteristics may vary between different regions or stages of tumor growth. Patients with multiple genetic alterations present additional challenges, as only a small percentage of advanced cancer patients have actionable mutations that can be targeted with existing drugs. Furthermore, gene expression profiling has not yet become a standard practice in clinical settings.
Conclusion
Precision oncology is rapidly advancing as a research field, leading to a multitude of new therapeutic options based on genomic and molecular biomarkers. The development of precision medicine targeting cancer has significantly increased the potential for cancer patients to extend their lifespan, improve their quality of life, and ultimately survive this devastating disease. This innovative approach to cancer treatment allows for targeted treatment of tumors, reducing side effects and damage to healthy cells while increasing the likelihood of treatment success. Despite the high cost and limited availability of precision medicine for all types of cancer currently, the goal is to eventually customize treatments based on the unique genetic and protein alterations in each individual’s cancer at a more affordable price.
References
[1] Alain Braillon1 and Adam Edward Lang; Eur. J. Epidemiol. 2023, 38, 391
[2] E. V. Jensen, et al; Natl. Cancer Inst. Monogr. 1971, 34, 55-70
[3] Stuart L. Rulten, et al; Int. J. Mol. Sci. 2023, 24, 12613
[4] Juliette Brownlie, et al; Curr. Opin. Pharmacol. 2023, 70, 102381
[5] Tong-Mei Shi, et al; J. Med. Chem. 2024, 67, 2238-2263
[6] Kailee A. Rutherford, et al; Mol. Cancer Ther. 2024, 23, 454-563
[7] Joaquin Mateo, et al; Nat. Med. 2022, 28, 658-665