Drug design is a sophisticated pharmaceutical discipline with a rich history of achievements. Since the late 19th century, when Emil Fisher proposed the concept of drug-receptor interaction as a key and lock mechanism, significant progress has been made in the field. Over time, drug design has evolved into a well-structured science with a solid theoretical foundation and practical applications. Today, it stands as the leading approach to drug discovery, leveraging advancements in science and technology to develop effective, specific, non-toxic, safe, and well-tolerated medications.
The cornerstone of any successful project lies in the meticulous design and thorough planning that take into account all potential factors that could impact its outcome. This principle is especially crucial in the realm of intelligent drug design, where the key to successful drug discovery and development lies in identifying molecules with beneficial characteristics. However, the process is highly complex, time-consuming, and resource-intensive, necessitating multi-disciplinary expertise and innovative approaches influenced by numerous critical factors. Without comprehensive drug design, there is a risk of failing to obtain FDA approval after investing substantial time and resources. Medicinal chemists play a pivotal role in conscientiously designing drug candidates, drawing upon their specialized knowledge and expertise to navigate the drug discovery process.
Several steps are involved in drug development, including:
- Discovery and development: Research for a new drug begins in the laboratory
- Preclinical research: Drugs undergo laboratory and animal testing to evaluate safety
- Clinical research: Drugs are tested on people to ensure safety and effectiveness
- FDA review: FDA review teams thoroughly assess all submitted data to determine approval
- FDA post-market safety monitoring: FDA monitors drug and device safety after products are available for public use
The process of drug discovery begins with the identification of a hit molecule, which demonstrates a desired activity in a screening assay. Subsequently, the structure of this molecule is refined to enhance affinity and selectivity, minimize toxicity, improve solubility in water and lipids, enhance general ADME properties, and transform the hit molecule into a lead molecule. Further optimization of the lead molecule culminates in the development of a drug candidate for preclinical trials.
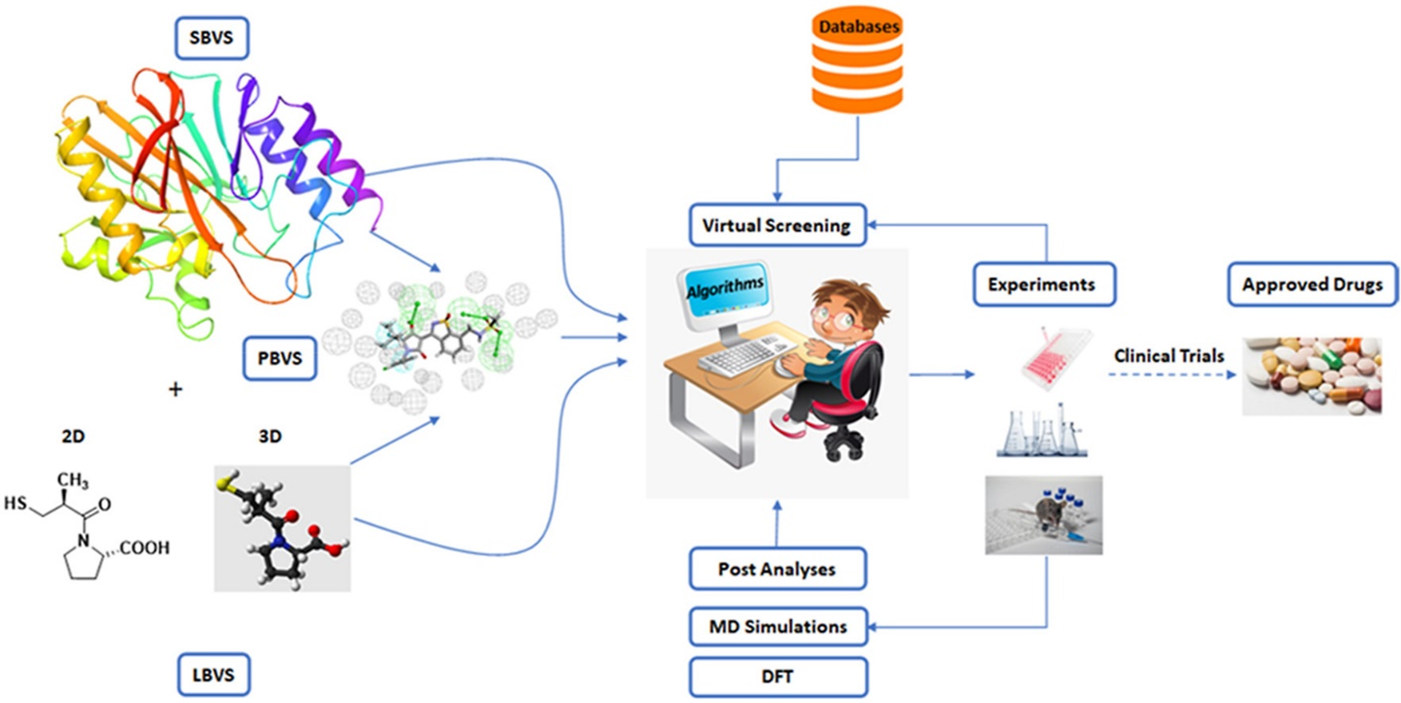
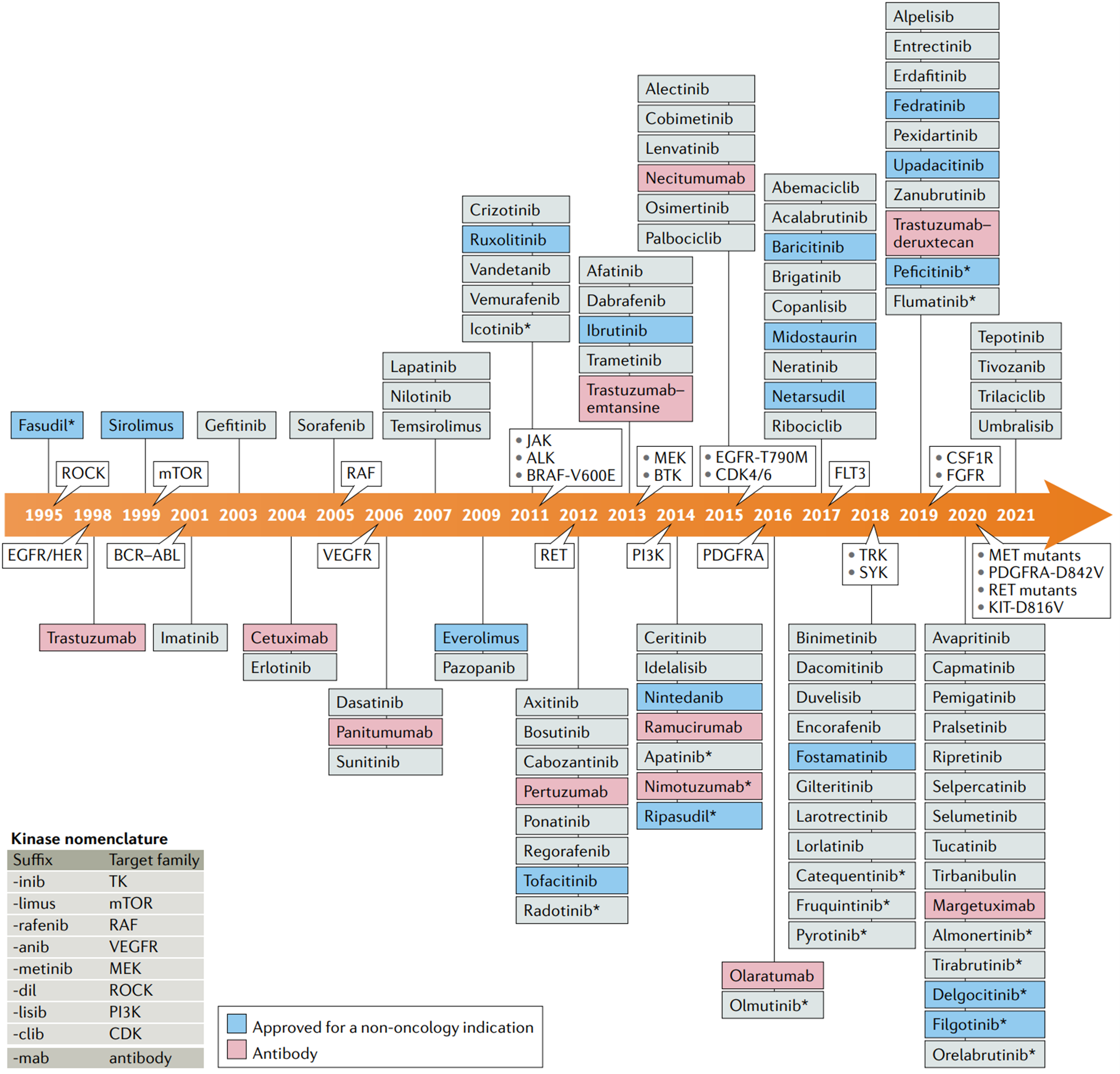
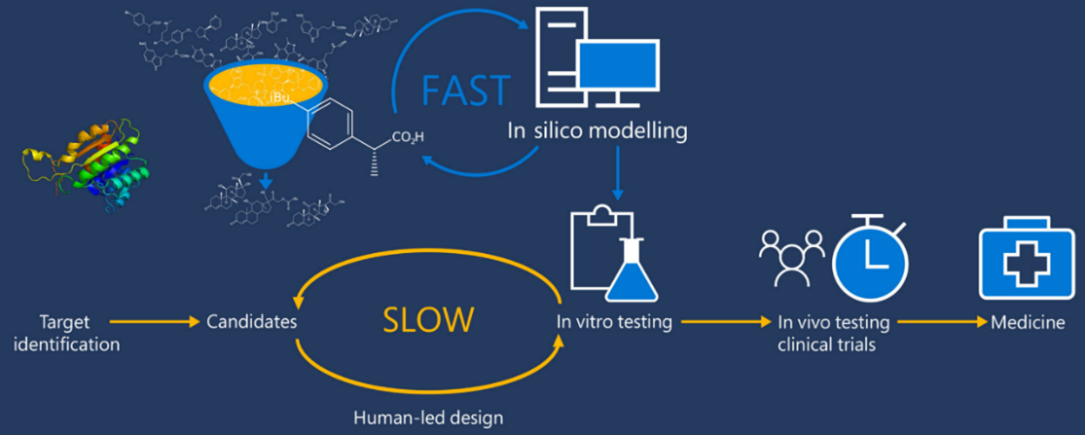
Medicinal chemists start drug design in the Discovery and Development phase. While advancements have been made in the modernization of the drug discovery process, most of the key factors for drug design have remained consistent. The integration of artificial intelligence (AI) and deep learning (DL) has recently accelerated the drug design process, improving efficiency and effectiveness. There are two main types of drug design: ligand-based drug design (LBDD) and structure-based drug design (SBDD). SBDD can be further categorized into de novo design and virtual screening, while LBDD can be categorized into QSAR, scaffold hopping, pseudo receptors, and pharmacophore modelling as illustrated in Figure 1 [1].
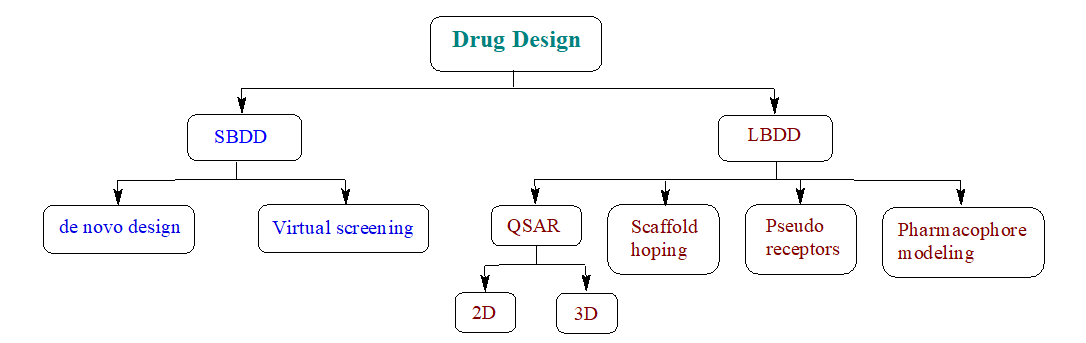
Figure 1: Types of drug design
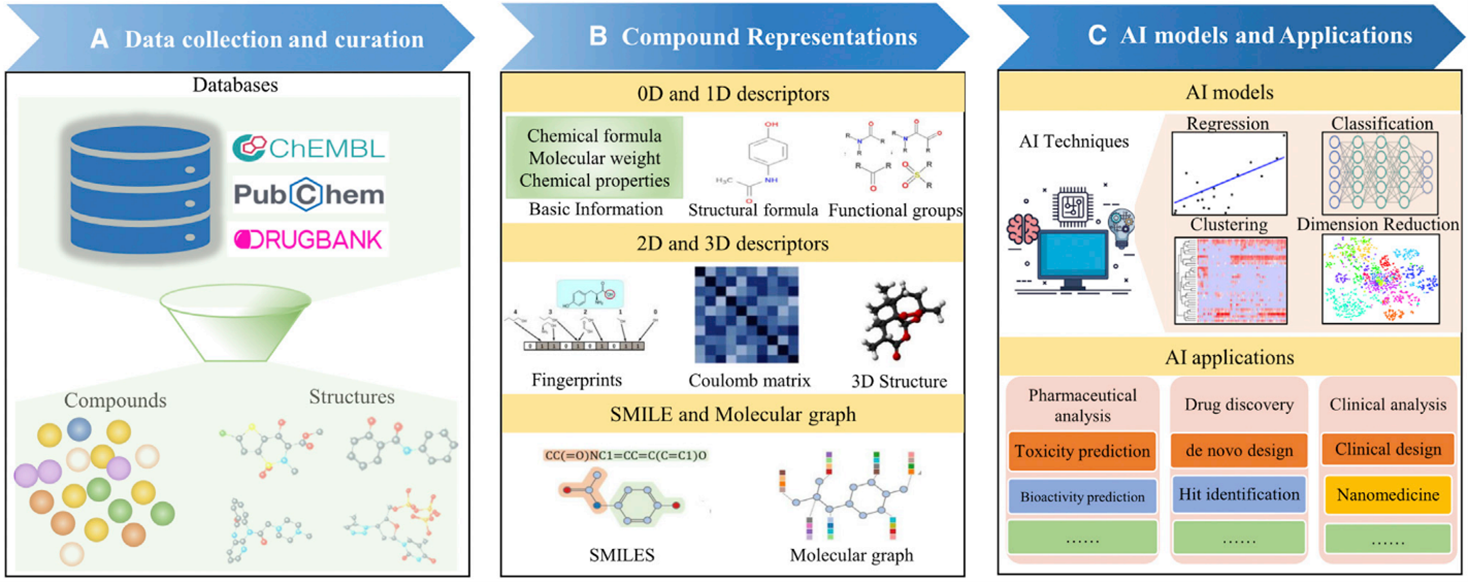
The elucidation of protein structures is achieved through various techniques such as X-ray crystallography, NMR spectroscopy, and cryogenic electron microscopy, with data being stored in the Protein Data Bank (PDB), which currently holds over 180 thousand structures. While the majority of these structures are single proteins in their apo-form, a subset includes complexes with ligands, providing valuable information on protein-binding sites. A significant advancement in modern drug design is the utilization of in silico modeling technologies for virtual screening, compound design, energy calculations, SAR and QSAR analysis, ADME modeling, and drug-target interaction modeling. To leverage these advanced technologies, molecular structures must be numerically encoded to enable analysis, search, visualization, and comparison. Encoding can be done using binary strings, smiles strings, 2D graphs, and 3D structures. The incorporation of 3D molecular modeling and visualization is recognized as a significant achievement in drug design technologies.
Computer-aided drug design (CADD) is widely recognized as a powerful tool in the drug discovery process. Techniques such as structure-based and ligand-based drug design through CADD provide crucial insights for molecular docking, molecular dynamics, and ADMET. Molecular docking is a computational method used to determine the precise binding pose of a protein-ligand complex and assess the strength of the interaction. AI and its subsets, Machine Learning (ML) and Deep Learning (DL), play a significant role in modern drug design by using vast datasets found in academic journals, patents, books, dissertations, reports, conferences, and clinical trials to generate novel and promising drug molecules.
The concept of sequence-to-drug represents an innovative approach in computational drug design that leverages protein sequence data through end-to-end differentiable learning. This cutting-edge methodology utilizes Transformer models such as CPI2.0 and AlphaFold as foundational tools, showcasing their ability to generalize across proteins and compounds. It is crucial to interpret the binding knowledge obtained from Transformer models to aid in the discovery of new hits for challenging drug targets and to identify new targets for existing drugs through reverse application. Unlike traditional drug design projects that involve a complex, human-engineered pipeline with independently optimized steps, this new concept streamlines the entire learning process in a self-consistent and data-efficient manner, potentially minimizing error accumulation in complex pipelines (Figure 2) [2].
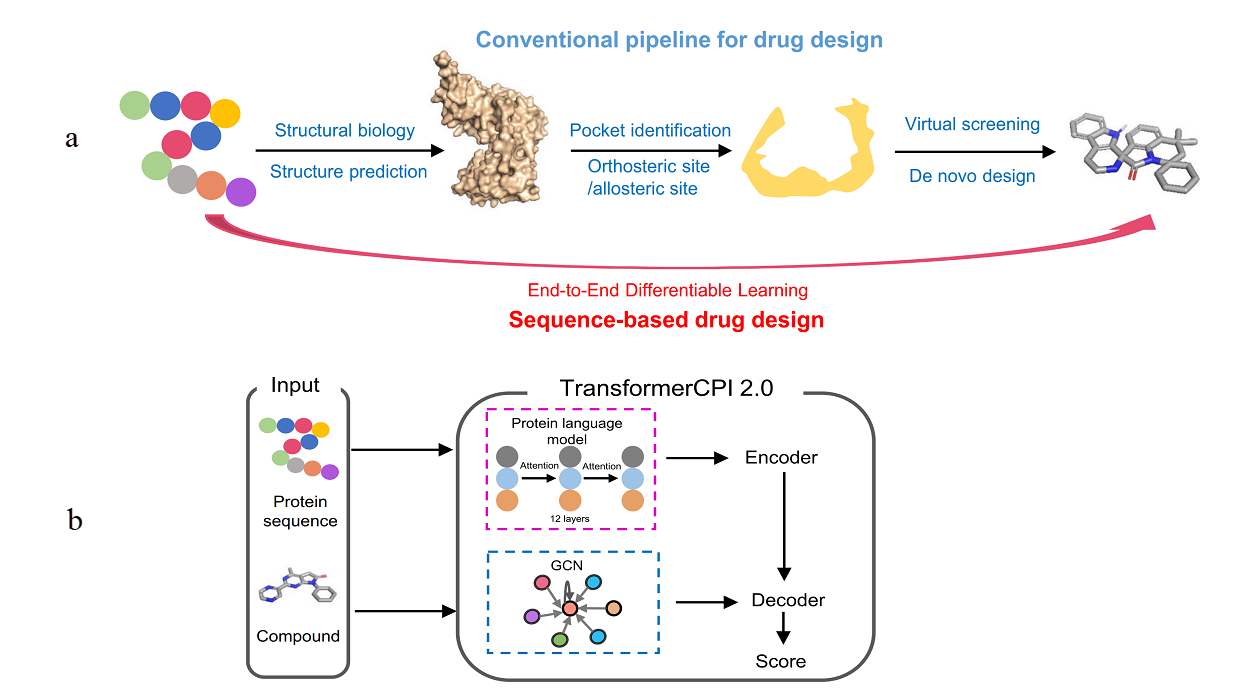
Figure 2: a) The conventional pipeline for target-based drug design and the sequence-to-drug concept; b) The computational pipeline of TransformerCPI2.0
The utilization of fragment linking as a valuable tool for the rational design of drug leads has been widely recognized in both academic and pharmaceutical settings. This approach is commonly employed to develop small molecule protein inhibitors and potential drug candidates. The initial stage of the fragment-based drug discovery process involves identifying fragments that exhibit weak binding affinity to the target protein, typically in the micromolar to millimolar range. Fragments are characterized as low molecular weight (<300 g/mol) and highly soluble organic molecules. Subsequently, these fragments are connected by a linker and optimized to create a single molecule with enhanced potency and improved drug-like properties. Two illustrative examples are provided in Figure 3 [3].
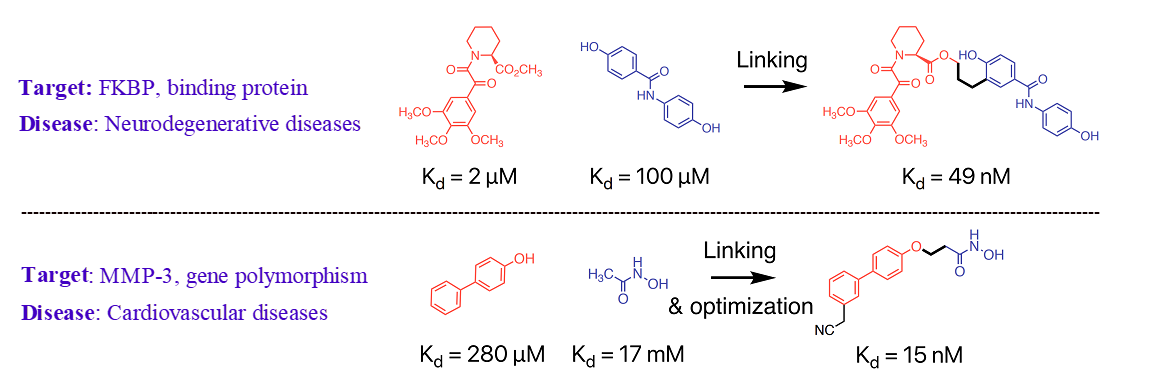
Figure 3: Fragment linking and optimization
In the field of drug discovery and development, Lipinski’s Rule of Five is a crucial consideration when designing small molecule drug candidates. This rule assists in predicting whether a biologically active molecule will have the necessary chemical and physical properties for oral bioavailability. The Rule of Five is based on specific physicochemical properties that impact pharmacokinetic drug properties such as absorption, distribution, metabolism, and excretion. According to Lipinski’s Rule of Five, orally active drugs generally should have no more than 5 hydrogen bond donors (HBDs), no more than 10 hydrogen bond acceptors (HBAs), a molecular mass less than 500 Da, and a partition coefficient (Clog P) not greater than 5. In drug design, molecules of interest generally exhibit a lower number of HBDs compared to HBAs. However, published data analyses for drug-like compounds do not definitively support the notion that HBDs pose greater challenges than HBAs in terms of ADME [4]
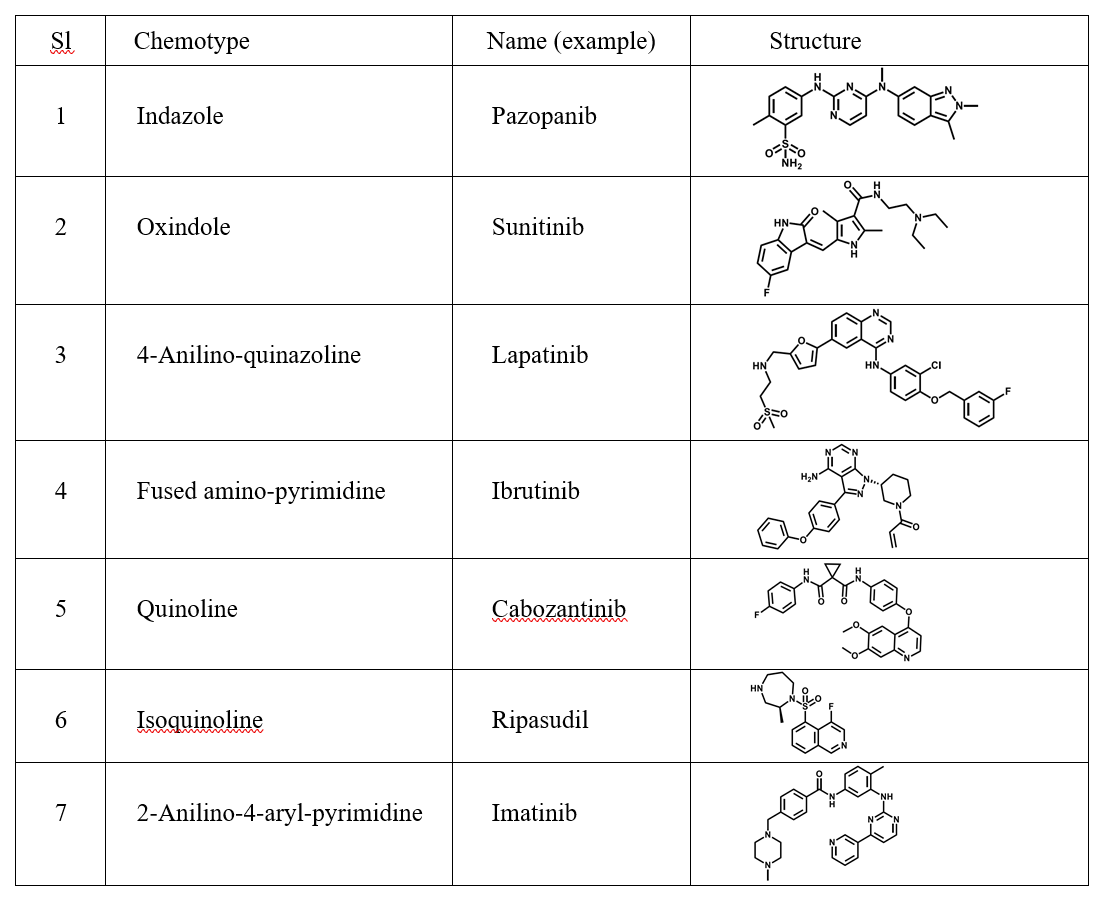
In the field of medicinal chemistry, bioisosteres are chemical substituents or groups that exhibit similar physical or chemical properties, resulting in comparable biological effects within the same compound. Utilizing bioisosteres in drug discovery offers a strategic and effective method to enhance molecule properties, such as increasing potency, addressing pharmacokinetic challenges, minimizing off-target effects, and optimizing physicochemical characteristics. The utilization of bioisosteres in refining bioactive compounds is a widely recognized and continually evolving approach that fosters innovation. For example, an illustration of bioisosteres related to carboxylic acid can be seen in Figure 4 [5].
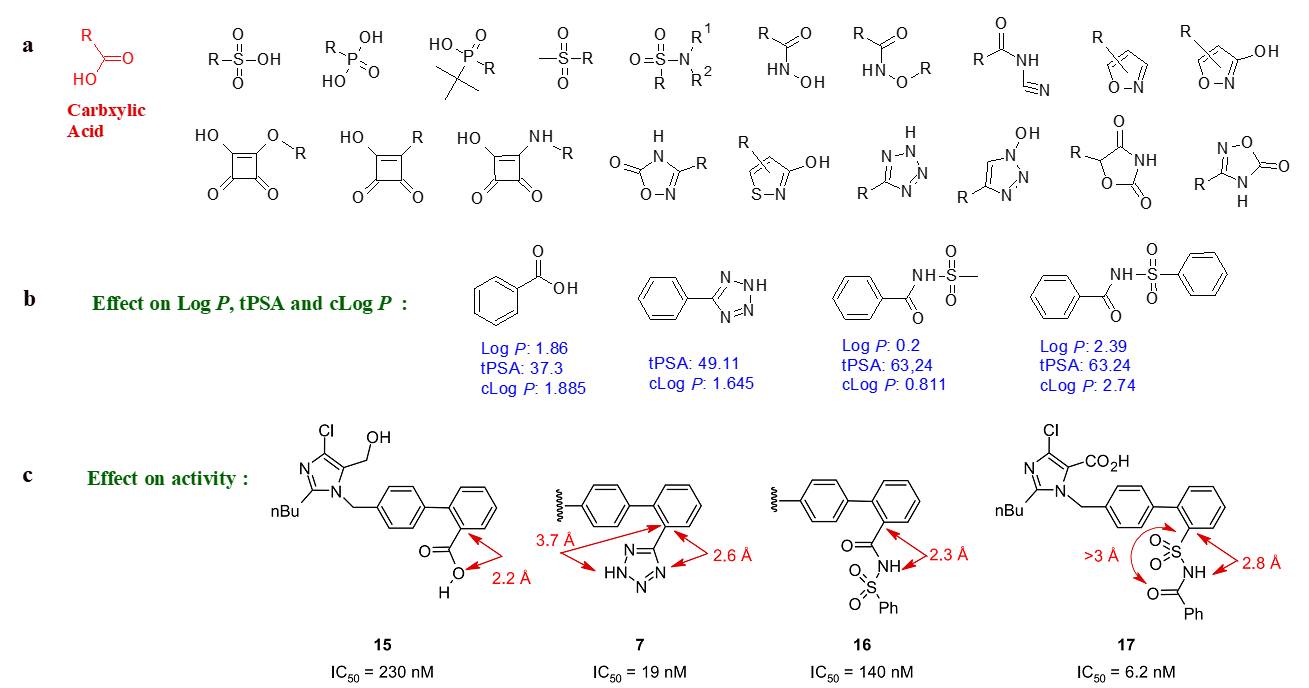
Figure 4: a) Examples of carboxylic acid bioisosteres; b) Bioisosteres: effect on properties; c) Bioisosteres: effect on bond length and activity
The process of drug design is a powerful tool within the pharmaceutical industry for discovering new drug leads against important targets. However, designing and synthesizing organic molecules with specific pharmaceutical applications presents a significant challenge, as it requires consideration of various factors such as selectivity, potency, pharmacokinetic properties, and scalability. It is worth noting that the majority of drug design approaches heavily rely on reported biological activity and properties data, which are often limited in quality and reliability. The drug design process is predominantly theoretical, which may result in discrepancies between expected and actual outcomes. Furthermore, while designing molecules may seem straightforward, their laboratory preparation can be quite challenging.
Innovative and cost-effective strategies are necessary for advancing drug discovery, including the utilization of artificial intelligence to efficiently analyze vast datasets for target identification and ligand selection. The ultimate objective of drug development is to create personalized drugs that are safe, effective, and rapidly developed. While this goal may seem ambitious, it is certainly achievable in the near future.
References:
[1] Layla Abdel-Ilah, Elma Veljović, et al; IJERT 2017, 6, 582-587
[2] Lifan Chen, Zisheng Fan Jie Chang, et al; Nat. Commun. 2023, 14, 4217
[3] Alexandre Bancet, Claire Raingeval, et al; J. Med. Chem. 2020, 63, 11420-1435
[4] Peter W. Kenny; J. Med. Chem. 2022, 65, 14261-14275
[5] Nicholas A. Meanwell; J. Agric. Food Chem. 2023, 71, 18087-18122