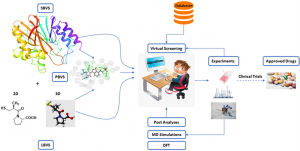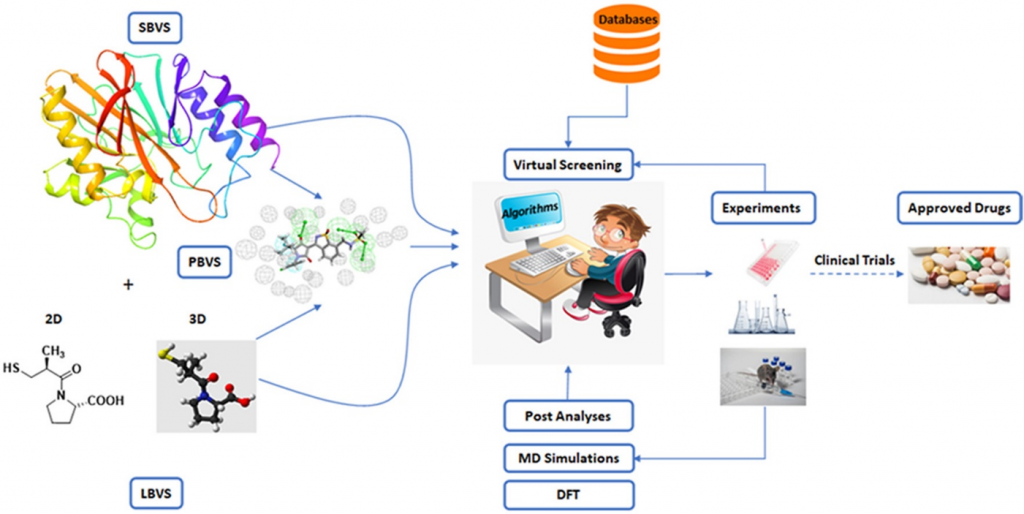
The process of drug discovery is an intricate, expensive, and time-consuming endeavor with a notably low success rate due to the complex nature of biological systems involving diverse heterogeneous elements that interact with each other, forming subsystems at different organizational levels. Consequently, extensive research has been conducted on molecular-level interactions within the context of disease mechanisms and drug discovery, resulting in the development of computational approaches. As a result, numerous databases provide readily available information on the relationships between drugs, diseases, proteins, and related concepts. Computational applications and tools have been widely adopted since the early 1980s to facilitate and expedite the discovery of drugs, giving rise to the field of computer-aided drug design (CADD). Over time, advancements in theoretical chemistry, physics, computer science, and statistics, coupled with the exponential growth in computational power, have greatly contributed to the field’s progress. Additionally, the evolution of data science has facilitated the transformation of extensive knowledge, enabling the emergence of bioinformatics and cheminformatics.
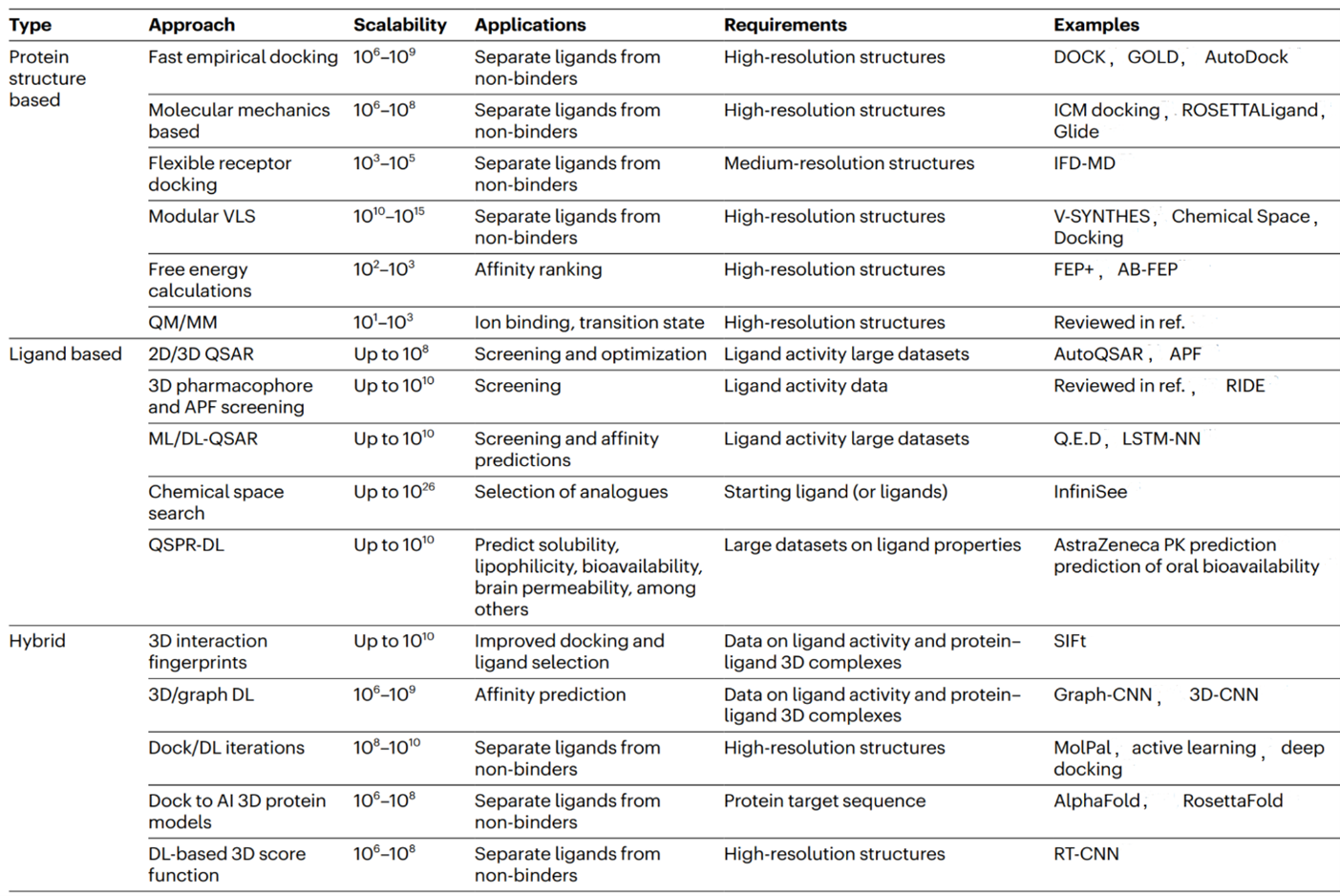
Computational methods are primarily employed in the early stages of drug discovery, where they play a crucial role in understanding disease biology, prioritizing drug targets, and optimizing chemical entities for therapeutic intervention. The main objectives of in silico approaches in drug discovery are to generate improved compounds with desirable in vitro and in vivo properties. Additionally, computational analysis aids in decision-making and guides experimental programs, reducing the number of candidate compounds that need to be evaluated experimentally. Presently, computational approaches have become indispensable tools at all stages of the drug discovery and development process. There are various computational methodologies available to assist researchers in identifying and investigating potential new drug candidates. Notably, structure-based drug design (SBDD) and ligand-based drug design (LBDD) are predominantly employed for drug design through CADD. Table 1 [1] provides a comprehensive overview of the primary computational approaches used to screen protein targets for potential ligands.
Table 1: Major types of virtual screening algorithms
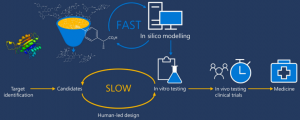
With the prolonged development period, increased cost, and limited probability of success, the rapid and effective formulation and synthesis of novel drug molecules pose significant challenges. The drug discovery process typically entails a design-make-test-analyze (DMTA) cycle lasting 10 to 15 years and incurring an average cost exceeding billion dollars to bring a new drug to the market. To mitigate these challenges and optimize time, resources, and risk, the incorporation of CADD methodology has become widespread. Research indicates that the utilization of CADD techniques can result in a 50% reduction in drug research and development costs. Additionally, the implementation of computationally driven drug discovery significantly shortens the identification time for potential drug candidates compared to traditional synthesis-driven discovery, as depicted in Figure 1 [1].
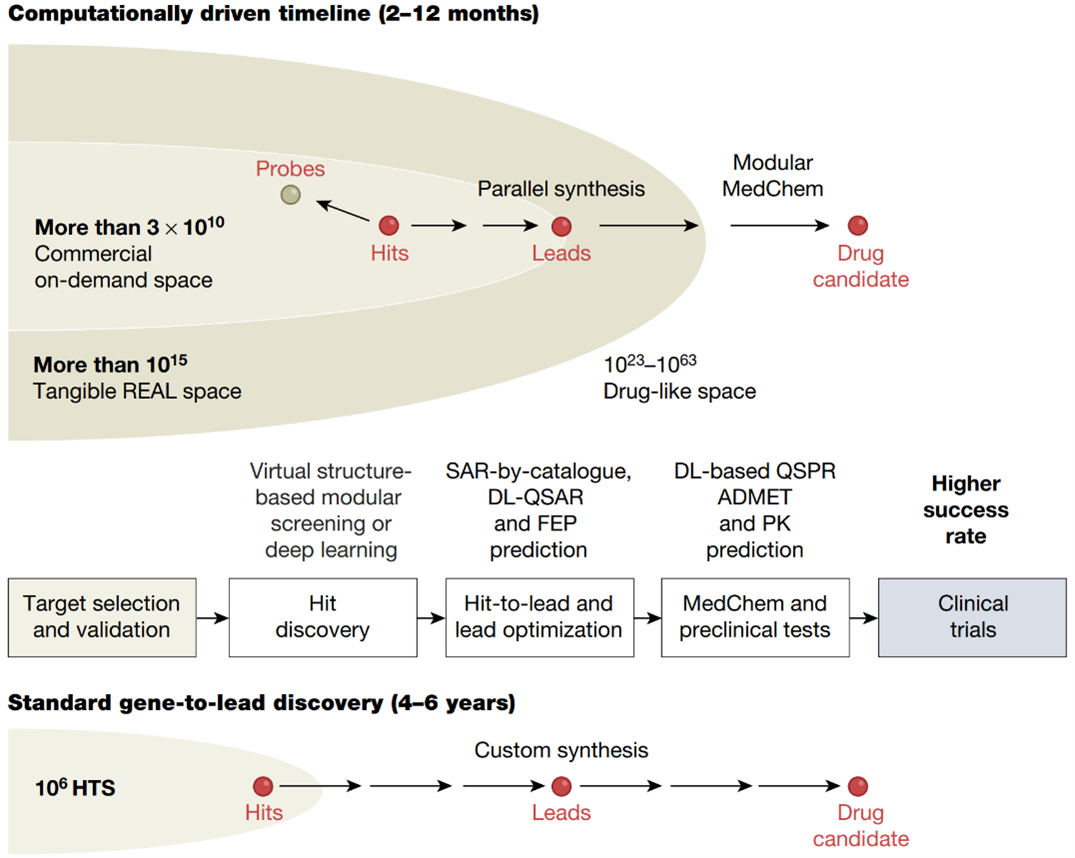
Figure 1. Computationally driven drug discovery. Schematic comparison of the standard HTS plus custom synthesis-driven discovery pipeline versus the computationally driven pipeline.
The field of drug discovery relies on several essential computational approaches, such as SBDD, LBDD, virtual screening, molecular docking, pharmacophore, quantitative structure-activity relationship (QSAR), and absorption, distribution, metabolism, excretion, and toxicity (ADMET).
- SBDD involves the calculation of the interaction or bio-affinity between tested compounds and proteins with known three-dimensional (3D) structure. This enables the design of therapeutic molecules with improved interactions with target proteins.
- LBDD, on the other hand, focuses on target proteins with unknown 3D structure but known ligands that bind to the intended target location. Through docking, these ligands can be used to create a pharmacophore model or molecule with the necessary structural characteristics to bind to the target active site. LBDD considers substances with similar structural similarities to have comparable biological actions and interactions with the target protein.
- Virtual screening has become a convenient tool for identifying bioactive compounds. It utilizes information about the protein target or known active ligands. There are two types of virtual screening approaches: structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS).
- Molecular docking (Figure 2) [2] is an in-silico approach used to predict the location of small molecules or ligands within the active region of their target protein. It accurately estimates the most favorable binding modes and bio-affinities of ligands with their receptors. Prediction of binding posture, bio-affinity, and virtual screening are the three primary goals of molecular docking, which are interconnected. The search and scoring algorithms used in molecular docking are fundamental tools for generating and evaluating ligand conformations.
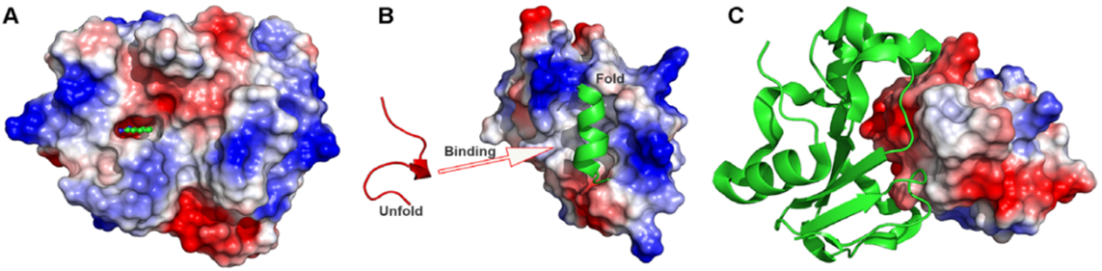
Figure 2. Molecular docking: (a) Small-molecule ligand binding, (b) peptide binding and (c) protein–protein interactions
- Pharmacophore refers to a schematic representation of bioactive functional groups and their interatomic distances.
- QSAR is a technique employed when structural-based approaches are not applicable due to a lack of knowledge about the target macromolecule structure. It provides information on the relationship between chemical structure and biological activity. The main advantage of QSAR is its ability to identify characteristics of new chemical compounds without the need for production and testing.
- ADMET evaluation of leads in the early stages of drug screening is necessary to address high attrition rates caused by poor pharmacokinetic profiles. Virtual screening can be utilized to filter hits and eliminate compounds with undesirable properties before comprehensive experimental testing. In silico ADMET filters, such as QSAR, use chemical or molecular descriptors to predict drug-like characteristics of compounds. ChemBioServer offers features such as displaying and graphing molecular characteristics, filtering compounds based on chemical quality, steric conflicts, and toxicity, searching for substructures, clustering compounds, and proposing a representative for each group.
The advancement of generative chemical spaces can be further facilitated by innovative computational methodologies in synthetic chemistry. For instance, the utilization of deep learning-based retrosynthetic analysis enables the prediction of novel iterative reaction sequences, synthetic routes, and feasibility. The discovery process of over 70 commercialized drugs included the use of computational techniques, as outlined in Table 2 [3] for example. It is important to note that in most cases, the initial drug lead (rather than the final commercial drug) was discovered with the aid of computer-aided drug design (CADD) techniques.
Table 2: Commercial drugs that made use of CADD during the discovery process
| Drug | Biological action | Computational contribution to drug discovery | Approval |
| Captopril | Angiotensin converting enzyme (ACE) | LBDD (Ligand-based drug design) and Structure-activity relationship (SAR) and SBDD (Structure-based drug design) | 1981 |
| Norfloxacin | Topoisomerase II, IV | SBVS, LBDD and QSAR Modelling | 1986 |
| Imatinib | Tyrosine kinase | SBDD (chemical libraries were screened for inhibitors against Bcr-Abl tyrosine kinase) | 1990 |
| Epalrestat | Aldose Reductase | MD and SBVS | 1992 |
| Cladribine | Adenosine deaminase | SBDD (VS and Docking) | 1993 |
| Saquinavir | HIV-1 protease | SBDD (Transition-state mimetic concept) | 1995 |
| Indinavir | HIV-1 protease | SBDD (Transition-state mimetic concept guided by molecular modelling and X-ray crystal structure) | 1996 |
| Zanamivir | Influenza Neuraminidase | SBDD (Computer-assisted modelling of the active site) | 1999 |
| Lopinavir | HIV-1 protease | SBDD (Transition-state mimetic concept) [3D modelling and docking. Energy minimization using DISCOVER CVFF force field] | 2000 |
| Eptifibatide | Glycoprotein IIb/IIIa | Peptide-based (barbourin) design | 2001 |
| Valsartan | Angiotensin II receptor | Superimposition of energy-minimized conformation and QSA | 2002 |
| Enfuvirtide | HIV-1 protease | Homology Modelling | 2003 |
| Erlotinib | EGFR kinase | SBVS | 2005 |
| Ambrisentan | Endothelin-A receptor | SBDD (Docking), FBDD and Virtual Screening | 2007 |
| Raltegravir | HIV-1 integrase | Combining MD with flexible-ligand docking | 2007 |
| Tomudex | Thymidylate synthase | SBDD | 2009 |
| Crizotinib | ALK and ROS1 | SBDD and SAR | 2011 |
| Rivaroxaban | Factor Xa | HTS, SBDD and Virtual SAR | 2011 |
| Dolutegravir | HIV-1 Integrase | PBDD (two-metal binding pharmacophore structural based design) | 2013 |
| Saroglitazar | PPAR | Combined virtual screening of 3D databases, SBDD and Pharmacophore Modelling | 2013 |
| Grazoprevir | NS3/4 A protease | Molecular Modelling and Docking-derived approach | 2016 |
| Rucaparib | Poly (ADP-ribose) polymerase (PARP-1) | Ligand-based molecular modelling | 2016 |
| Acalabrutinib | Bruton’s tyrosine kinase | SAR, SBDD and Docking | 2017 |
| Betrixaban | Serine protease Factor Xa (fXa) | Molecular Docking | 2017 |
| Brigatinib | ALK | Docking and Homology Modelling | 2017 |
| Copanlisib HCl | Phosphoinositide 3-kinase (PI3K) | SBDD (Xray crystallography and Docking) and LBDD (based on lead scaffold) | 2017 |
| Vaborbactam | β-Lactamase | Docking and MD | 2017 |
| Duvelisib | PI3K Kinase | SBDD (Molecular docking, virtual screening) and LBDD (lead optimization and SAR) | 2018 |
| Apalutamide | Androgen receptor inhibitor | SBDD and SAR | 2018 |
| Dacomitinib | Oral kinase | Combined FBDD and SBDD | 2018 |
| Talazoparib Tosylate | Poly (ADP-ribose) polymerase-PARP | SBDD, SAR and Lead Optimization | 2018 |
| Darolutamide | Androgen receptor | SBDD (Docking and MD) | 2019 |
| Erdafitinib | FGFR tyrosine | Combined FBDD and SBDD | 2019 |
| Fedratinib HCl | Tyrosine kinase | SBDD (Virtual screening and Molecular Docking) | 2019 |
| Selinexor | Nuclear export | SBDD (consensus induced fit docking | 2019 |
| Zanubrutinib | Bruton’s tyrosine kinase inhibitor | Combined FBDD and SBDD | 2019 |
The profound impact of computer-aided drug design (CADD) on pre-clinical drug development is evident. Furthermore, the application of CADD is continuously expanding alongside advancements in virtual screening and molecular docking. The integration of molecular dynamics simulations and experimental evaluation has the potential to enhance the reliability of refining computational models for identifying promising prospective hits. It is important to highlight that computational approaches continue to be a crucial resource in pre-clinical drug development, with a proven track record in facilitating the development of new drugs and repurposing existing ones.
Despite the numerous advancements and applications in drug discovery and development utilizing computational approaches, there are still several unresolved obstacles and challenges. These include the lack of synergistic computational models, quality datasets, standardization, accurate scoring functions, issues with multi-domain proteins, and assessment of multi-drug effects. Additionally, improvements in specific areas are necessary, such as enhancing the efficiency of virtual screening, increasing the quantity and quality of online computational resources, further advancing computational chemogenomics, designing drugs for multiple molecular targets, enhancing predictive toxicity models and side effects, and collaborating with other disciplines to optimize the search for bioactive compounds for treating and preventing diseases.
References
[1] Anastasiia V. Sadybekov and Vsevolod Katritch; Nature 2023, 616, 673-685.
[2] Jinan Wang, et al; QRB Discovery 2022, 3: e13, 1–12.
[3] Victor T. Sabe, Thandokuhle Ntombela, et al; Eur. J. Med. Chem. 2021, 224, 113705.