Autophagy-Driven Approaches in Drug Development
Autophagy is a fundamental cellular degradation process responsible for the removal of damaged or expendable proteins, aggregates, and droplets by transporting them to lysosomes. It serves as a protective mechanism for cells and organisms in response to stress. Research in human genetics and pathophysiology has shown that disruptions in autophagy can impact cellular homeostasis and disease development. Dysregulated autophagy is associated with a variety of diseases, including neurodegenerative disorders, infectious diseases, autoimmune conditions, and cancer. The significance of autophagy for human health was recognized in 2016 when the Nobel Prize for Medicine or Physiology was awarded to Professor Yoshinori Ohsumi for his groundbreaking work on elucidating the mechanisms of autophagy. The molecular components of autophagy are currently under investigation as potential targets for drug development and therapeutic intervention of various diseases.
Autophagy is a complex process that is regulated by multiple signaling pathways, such as Jun N-terminal kinase, GSK3, ERK1, Leucine-rich repeat kinase 2, PTEN-induced putative kinase 1, and parkin RBR E3. Protein kinases play a crucial role in the regulation of autophagy, either activating or inhibiting the process. There are three main subtypes of autophagy: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy or simply autophagy. Autophagy relies on a series of dynamic membrane events. It starts with the sequestration of cytoplasmic components by the isolation membrane, which forms the phagophore. Upon complete sealing, an autophagosome is formed, which later fuses with the lysosome to create an autophagolysosome. Lysosomal hydrolases degrade the inner membrane of the autophagosome, cytoplasmic constituents, and protein aggregates. The breakdown molecules, such as amino acids and nucleosides, are recycled as chemical energy or building blocks for other cellular processes. Various autophagy-related (Atg) proteins are involved in different stages of autophagy, either individually or in combination. Molecular mechanism of autophagy has been described in Figure 1 [1].
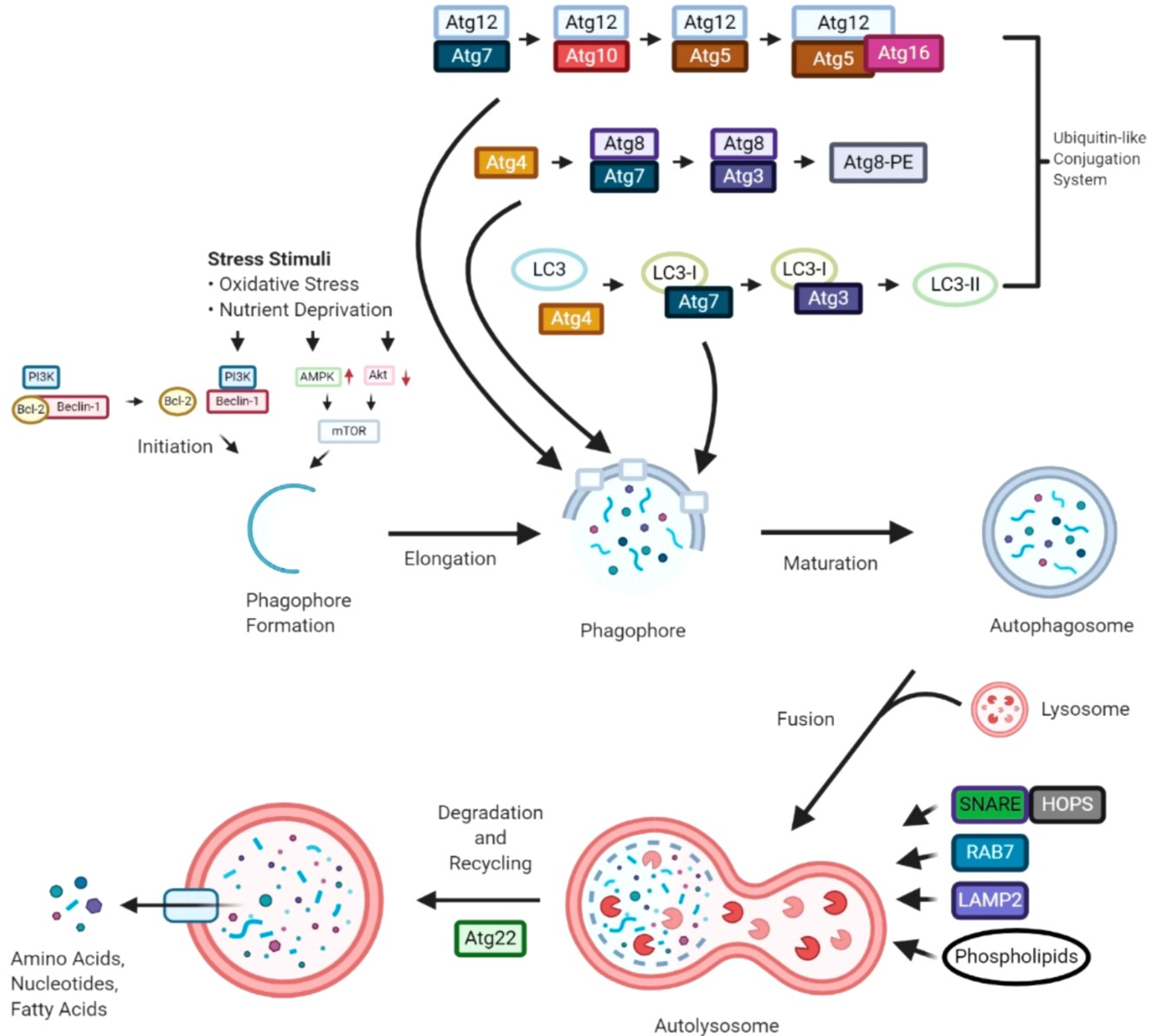
Figure 1: Molecular mechanism of autophagy
The importance of addressing autophagy dysfunction in a variety of human pathologies highlights the urgency of discovering new agents that can target autophagic genes and pathways in pathological conditions. The development of pharmacologic agents that can induce or inhibit autophagy has the potential to prevent the occurrence, delay the progression, and decrease the mortality of certain diseases. Strategies for autophagy-based drug design can be categorized into three main areas based on pharmacological mechanisms for therapy: autophagy inhibition, activation, and regulation to address diseases. Targeting specific cell populations through pharmacological inhibition of autophagy is a key therapeutic goal in the first scenario, with the effectiveness and therapeutic value of such interventions dependent on the stage of the autophagic cascade being targeted. Designing clinically useful autophagy activators in the second scenario requires a comprehensive understanding of the autophagic defects associated with each disease, as these defects can contribute to disease progression through various mechanisms. Furthermore, in some cases, autophagy alterations may not be directly linked to the primary disease etiology, but autophagy activation can still support compensatory mechanisms. Overall, autophagy-based therapeutic interventions hold promise but are complex and require careful consideration.
Autophagy is intricately regulated at the transcriptional level by transcription factors such as MITF, FOXO families, CREB, and ATF. Additionally, post-translational modifications play a crucial role in modulating autophagy, allowing for both positive and negative regulation through pharmacological interventions (Figure 2) [2]. For instance, mTORC1, a complex of mammalian target of rapamycin, inhibits autophagy, making mTOR inhibitors a common strategy to stimulate autophagy. Other pathways, such as mTORC2 and CMA, have also been associated with autophagy regulation. Various drugs, including ULK1 and ULK2 inhibitors, VPS34 inhibitors, and BH3 mimetics like Venetoclax, can inhibit autophagy. Nonpharmacological interventions like caloric restriction and exercise also play a role in inducing autophagy. The process of autophagosome membrane elongation involves ubiquitin-like conjugation systems, where proteins like ATG12 and ATG5 are conjugated in a complex enzymatic process. The LC3-PE conjugation, facilitated by ATG4B, is a critical step in autophagy and serves as a common marker for the process. The final fusion of autophagosomes with lysosomes, mediated by proteins like STX17, is essential for the degradation of autophagic contents. Lysosomal inhibitors like chloroquine, hydroxychloroquine, and Bafilomycin A1 can block this fusion process. Following fusion, lysosomal hydrolases break down the autophagic contents into amino acids, nutrients, and lipids, which are then utilized for various cellular processes, including protein synthesis and metabolism.
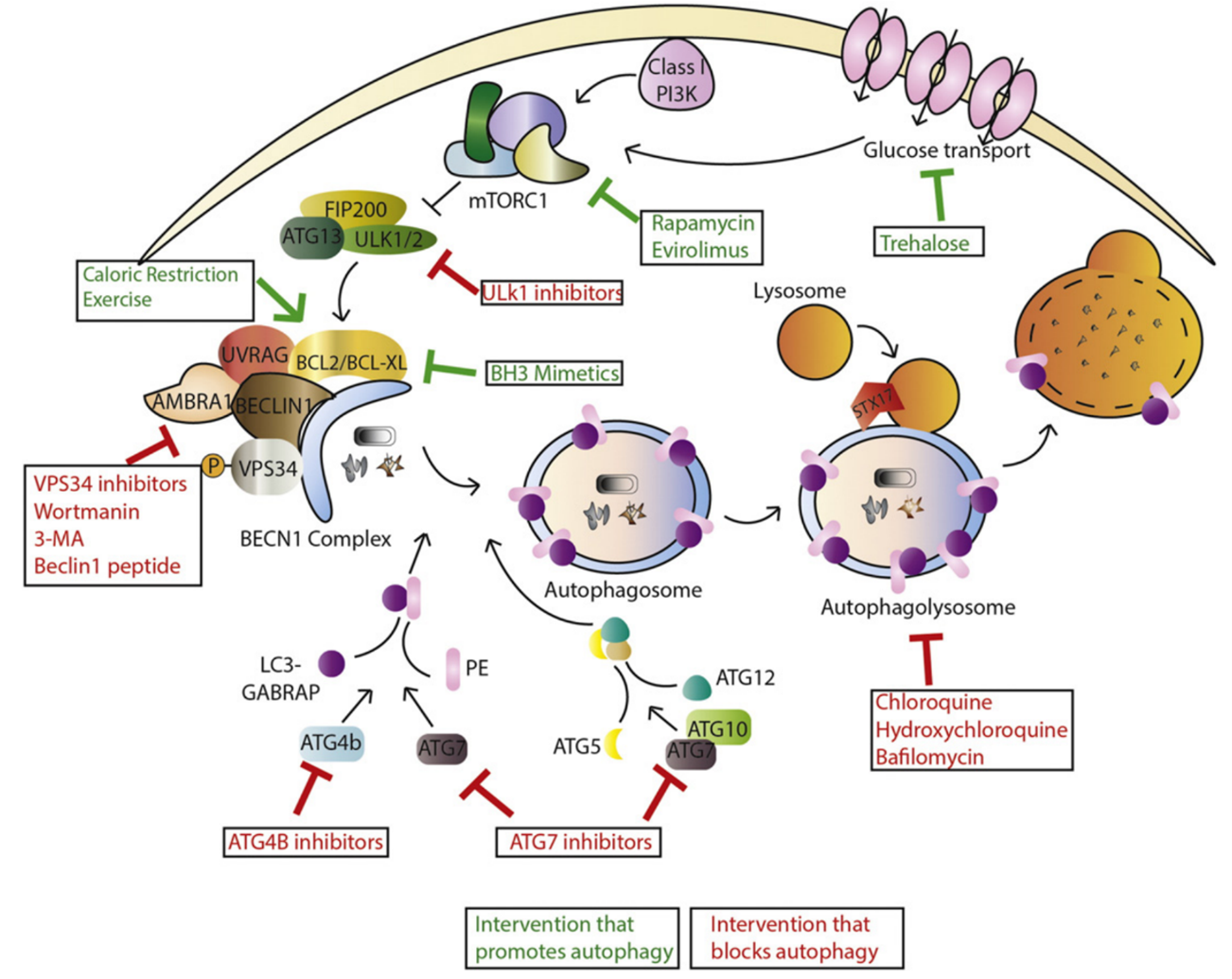
Figure 2: Interventions that target autophagy.
The activation of autophagy using small molecules has traditionally involved inhibiting mTORC1 or activating AMPK. Inhibition of mTORC1 prevents the phosphorylation of ATG13, ULK1, and ULK2 in the ULK1 complex, allowing for the activation of ULK1 by AMPK and increasing autophagy levels. AMPK can also phosphorylate RAPTOR, a component of mTORC1, reducing mTORC1’s suppression of autophagy. Rapamycin and rapalogs induce autophagy by forming a complex with FK506-binding protein (FKBP12), acting as an allosteric inhibitor of mTORC1. Compounds like Torin-1 selectively inhibit mTORC1’s kinase activity. Additionally, there are compounds that appear to induce autophagy independently of mTORC1. While the autophagy-inducing properties of these compounds are well known, identifying their molecular targets is crucial to confirm their potential as tool compounds. For example, Carbamazepine induces autophagy by reducing IP3 levels, resulting in decreased mitochondrial Ca2+ uptake, slightly impaired respiration, and AMPK activation. Felodipine inhibits cellular Ca2+ uptake, reducing calpain activity, a negative regulator of autophagy. Since both drugs are already approved, there is potential to repurpose them as autophagy inducers. A number of compounds are highlighted in Figure 3 [3], which have been reported to as autophagy inducers, inhibitors of autophagy initiation and inhibitors of autophagosome maturation and lysosomal activity.
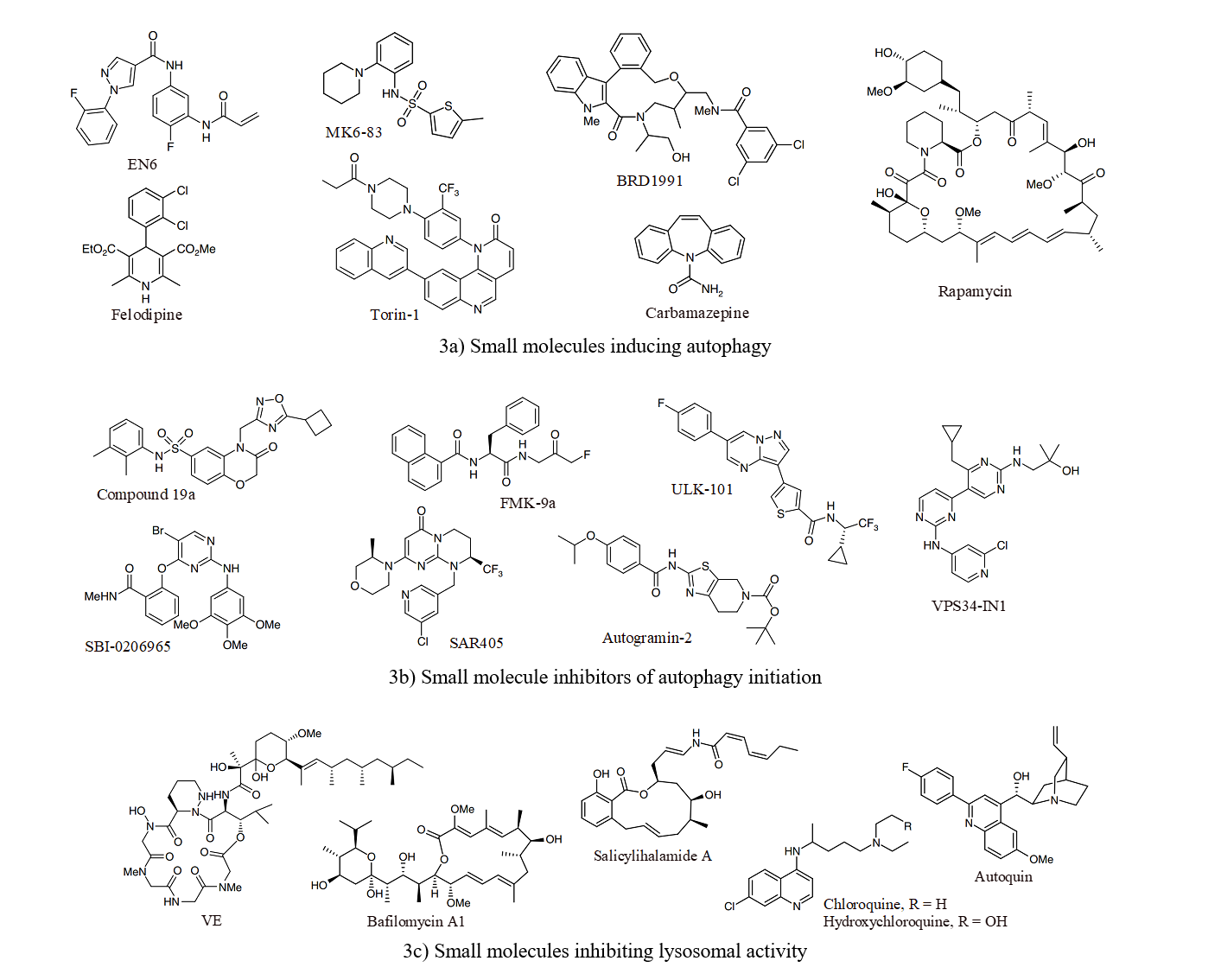
Figure 3: Induction of autophagy using small molecules
The dysfunction of autophagy can lead to disturbances in cellular and organismal homeostasis, potentially contributing to various human diseases including cancer, metabolic disorders, neurodegenerative diseases, aging, cardiovascular diseases, skeletal diseases, and reproductive diseases.
Autophagy and cancer: The autophagic pathway plays a significant role in colorectal carcinogenesis, with a dual impact on cancer development. Initially, autophagy promotes cancer suppression during the early stages of tumorigenesis but switches to protecting tumors during progression. Several autophagy-related genes and proteins act as tumor suppressors, with their inhibition leading to genomic instability and tumor initiation. Conversely, autophagy also supports tumorigenesis through mechanisms such as inhibiting p53 activation, suppressing antitumor immune responses, and maintaining redox and metabolic homeostasis. Deletion or mutation of key autophagy-related genes can impact tumor initiation, with overexpression of ATG16L2 reported in various cancers [4]. Small-molecule compounds targeting autophagy have been developed for cancer treatment, with ongoing clinical trials exploring their efficacy. Autophagy inhibitors are commonly used as anticancer agents in combination with chemotherapy, while autophagy activators require further investigation and testing in clinical settings.
Autophagy in cardiovascular diseases: Autophagy plays a critical role in maintaining cardiovascular health by supporting the function of cardiac myocytes, essential cellular components of the cardiovascular system. This process helps these cells manage harmful components and sustain physiological functions. As a result, autophagy is crucial for preventing various cardiovascular diseases, such as cardiomyopathy, ischemia-reperfusion injury, diabetic cardiomyopathy, cardiac hypertrophy, atherosclerosis, coronary artery disease, arrhythmia, chemotherapy-induced cardiotoxicity, and heart failure.
Autophagy in neurodegenerative diseases (NDs): NDs are characterized by the misfolding, aggregation, and accumulation of proteins, leading to cellular dysfunction, synaptic loss, and brain damage. Autophagy has been identified as a critical factor in the pathogenesis of NDs for two primary reasons. Firstly, deficiencies in autophagy have been associated with neurodegeneration. Secondly, the presence of toxic protein aggregates that can be cleared through autophagy has been implicated in diseases such as Alzheimer’s Disease (AD), Parkinson’s Disease (PD), and Huntington’s Disease (HD). Autophagy, a major degradation pathway, plays a crucial role in removing protein aggregates and damaged organelles in neuronal cells. As such, the development of drugs that target autophagy for the treatment of NDs and the investigation of the relationship between NDs and autophagy are essential areas of research.
The crucial role of autophagy in a variety of illnesses and disorders has spurred increased attention on drug discovery and development targeting autophagy. Over the last decade, both academic institutions and pharmaceutical companies have dedicated substantial resources to identifying effective drug candidates that manipulate autophagy for disease treatment. Autophagy presents a promising therapeutic target that can be pharmacologically manipulated at multiple stages. Currently, there are numerous autophagy-targeted compounds in various stages of development, with over 200 in preclinical studies and more than 100 in clinical trials. Combining autophagy modulators with existing drugs or adjuvant therapies has shown potential in improving treatment outcomes for various diseases, particularly in enhancing anti-tumor efficacy. While autophagic activators and inhibitors offer promise in disease treatment, challenges such as poor target selectivity, off-target toxicity, and drug resistance impede their straightforward clinical application. The discovery of small-molecule activators of autophagy for treating human diseases is still in its early stages, requiring further research and is likely to yield significant advancements and breakthroughs.
References
[1] Xin Chien Lee, Evelyn Werner and Marco Falasca; Cancers 2021, 13, 1211
[2] Christina G. Towers and Andrew Thorburn; EBioMedicine 2016, 14, 15-23
[3] Thomas Whitmarsh-Everiss and Luca Laraia; Nat. Chem. Biol. 2021, 17, 653-664
[4] Mengjia Jiang, Wayne Wu, et al, Eur. J. Med. Chem. 2024, 267, 116117
[5] Waleska Kerllen Martins, et al; Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100033 (for top Figure)